
Die MDR legt in Artikel 120 die Übergangsbestimmungen einschließlich der Übergangsfristen fest. Allerdings sind diese Übergangsbestimmungen und Übergangsfristen sehr komplex formuliert. Daher laufen die Hersteller Gefahr, sie falsch zu verstehen und regulatorische Anforderungen nicht zu erfüllen oder unnötige Aufwände zu betreiben.
Ein Ablaufdiagramm in Kapitel 2 dieses Artikels fasst die regulatorischen Anforderungen zusammen und schafft Klarheit. Eine Zusammenfassung steht auch als kostenloser Download zur Verfügung.
Dieser Beitrag berücksichtigt die im Februar 2023 vom EU-Parlament verabschiedete weitere Verschiebung der MDR-Übergangfristen.
1. MDR-Übergangfristen: Einführung
Die Diskussion um die Übergangsfristen beschränkt sich meist auf die Frage, wie lange ein Produkt in den Verkehr gebracht werden darf. Dabei kennt die MDR eine Vielzahl von Übergangsfristen.
Hersteller sollten bei den Übergangsfristen unter anderem die folgenden Aspekte unterscheiden:
Dieser Artikel verschafft einen Überblick über diese Übergangsfristen.
Unser Regulatory-Affairs-Experte Luca Salvatore schafft in dieser Podcast-Episode Klarheit, zeigt aber auch, welche Fragen noch nicht beantwortet sind.
Diese und weitere Podcast-Episoden finden Sie auch hier.
2. MDR-Übergangsfristen im Überblick
a) Zusammenfassung
| Aspekt | Übergangsfrist | Kommentar |
| Inverkehrbringung von Legacy-Produkten | siehe Ablaufdiagramm (Abb. 1) | Es geht um die erstmalige Bereitstellung |
| Weitere Bereitstellung und Inbetriebnahme von Legacy-Produkten | — | Die Einschränkungen („Sell-Off-Regelung“) sind mit der Änderungsverordnung weggefallen. |
| OEM-PLM-Konstrukte | Für diese Produkte gelten die gleichen Übergangsfristen. | Dem PLM muss die Technische Dokumentation vorliegen, um die Forderungen der MDR an die Post-Market-Surveillance sowie die Vigilanz erfüllen zu können. |
| „Verantwortliche Person“ / PRRC | — | Wird nicht benötigt für Hersteller von Legacy-Produkten. Lesen Sie hierzu den ausführlichen Absatz „Übergangsfristen“ im Artikel zur PRRC. |
| Post-Market-Surveillance | 26.05.2021 | Mit Einschränkungen bezüglich der EUDAMED; keine Einschränkung bei Überwachung und Berichten (z.B. PSUR) |
| Vigilanz | 26.05.2021 | Mit Einschränkungen bezüglich der EUDAMED |
| QM-System | 26.05.2021 (MDR-Produkte) 26.05.2024 (Legacy-Produkte) |
Die Änderungsverordnung verlangt ein MDR-Artikel-10-konformes QM-System für Hersteller von Legacy-Produkten. |
| UDI | siehe Tabelle unten | |
| Klinische Prüfungen | 26.05.2021 | Eingeleitete Prüfungen dürfen fortgeführt werden, allerdings gelten neue Meldepflichten. |
Der Artikel 10 der MDR findet bei Produkten, die von der Übergangsfrist profitieren, keine Anwendung. Davon explizit ausgenommen sind die o.g. Anforderungen u.a. zur Post-Market-Surveillance und zur Vigilanz.
Die Zusammenfassung finden Sie in diesem Merkblatt zum Download:
Zudem haben wir Ihnen eine konsolidierte Version der Artikel zu den Übergangsfristen erstellt.
Im August 2023 hat auch die EU auf ihrer Webseite ein Ablaufdiagrammpubliziert.
b) Inverkehrbringung, Bereitstellung und Inbetriebnahme
Wie lange Hersteller ihre bestehenden Produkte unter den alten Richtlinien (MDD/AIMDD) noch in den Verkehr bringen, bereitstellen und in Betrieb nehmen (lassen) dürfen, hängt u.a. von der Klasse der Produkte und der Gültigkeit möglicher Bescheinigungen ab.
Falls Sie noch Fragen zum Umstieg auf die MDR haben, nutzen Sie gerne das kostenlose Micro-Consulting des Johner Instituts.
c) EUDAMED, Registrierung
| Erste 6 Monate nach Veröffentlichung | Die nächsten 18 Monate danach | Danach | |
| MDR-Produkte und Legacy-Produkte | Keine Anforderungen | Alle Anforderungen außer Registrierung von Produkten (außer Sonderanfertigungen) (Artikel 29(4)) Bescheinigungen (Artikel 56(5)) (s. Artikel 123(3)e)) |
Alle Anforderungen |
Für Wirtschaftsakteure mit Sitz in Deutschland gelten gemäß dieser Veröffentlichung im Bundesanzeiger gesonderte Übergangsbestimmungen. Demnach müssen sich Hersteller und deren Bevollmächtigte bereits seit dem 26.05.2021 direkt in EUDAMED registrieren und nicht in der nationalen Datenbank des BfArMs, dem DMIDS. Gleiches gilt für Importeure von MDR-konformen Medizinprodukten.
d) Übergangsfristen für die UDI
Der Zeitpunkt, zu dem der UDI-Träger aufgebracht werden muss, hängt von der Klasse des Produkts ab:
| Klasse | Zeitpunkt |
| I | 26.05.2025 |
| IIa | 26.05.2023 |
| IIb | 26.05.2023 |
| III und implantierbare Produkte | 26.05.2021 |
Bei wiederverwendbaren Produkten, bei denen der UDI-Träger auf dem Produkt selbst zu platzieren ist, gewährt die MDR zwei zusätzliche Jahre.
Diese Übergangsfristen bezüglich UDI gelten jedoch nicht für Legacy-Produkte, also Produkte „which are compliant with the Medical Device Directives (MDD and AIMDD) and placed on the market after the application date of the Regulations“. Hierfür gibt es zwei weitere Dokumente zu beachten:
Im FAQ schreibt die EU:
In order to facilitate the transition to the new system, the new Regulations give manufacturers the possibility to place products on the market after the general application dates of the new
Regulations (and until 26 May 2024 at the latest) by virtue of valid Directive certificates. These legacy devices are not subject to UDI obligations but they should be registered in the Eudamed database. Timelines for registration as described under question 6 also apply to these products. More information on the operational aspects of the registration of legacy devices is available at the MDCG 2019-5 guidance document.FAQ der EU
e) Übergangsfrist für die Verantwortliche Person
Wenn Hersteller keine Produkte unter der MDR in den Verkehr bringen, sondern nur „Legacy-Produkte“ nach Art 120 (3), so gilt nach MDCG 2021-25 der Artikel 15 der MDR noch nicht. Dies bedeutet, dass diese Hersteller noch keine Verantwortliche Person benennen müssen.
Bisher hatten wir und auch viele andere dies auf andere Art interpretiert. Der Grund dafür war, dass die Pflichten für die Post-Market Surveillance und die Vigilanz von allen Herstellern ab dem 26.05.2021 erfüllt sein müssen. Die Verantwortliche Person ist für einen wirksamen Post-Market Surveillance- und Vigilanz-Prozess verantwortlich. Die Leitlinie MDCG 2021-25 betrachtet diesen Aspekt auch, kommt aber zu dem Schluss, dass Artikel 15 noch nicht von Herstellern von Legacy-Produkten anwendbar ist.
Alle Hersteller müssen in jedem Fall wirksame Prozesse für die Post-Market Surveillance und Vigilanz implementieren, nur die Überwachung durch die Verantwortliche Person ist noch nicht zwingend vorgeschrieben für Hersteller von Legacy-Produkten.
Details finden Sie auch im Artikel zur PRRC im Abschnitt „Übergangsfristen“.
3. Die MDR-Artikel 120 bis 123
a) MDR Artikel 120 Absatz 2
Originaltext
Der Artikel 120 trägt den Titel „Übergangsbestimmungen“. Er nennt darin auch die Übergangsfristen. Besonders relevant ist der zweite Absatz:
Bescheinigungen, die von Benannten Stellen vor dem 25. Mai 2017 gemäß den Richtlinien 90/385/EWG und 93/42/EWG ausgestellt wurden, bleiben bis zu dem in der Bescheinigung angegebenen Zeitpunkt gültig, außer im Fall von Bescheinigungen gemäß Anhang 4 der Richtlinie 90/385/EWG bzw. gemäß Anhang IV der Richtlinie 93/42/EWG, die spätestens am 27. Mai 2022 ihre Gültigkeit verlieren.
Bescheinigungen, die von Benannten Stellen ab dem 25. Mai 2017 gemäß den Richtlinien 90/385/EWG und 93/42/EWG ausgestellt wurden, die am 26. Mai 2021 noch gültig waren und die anschließend nicht zurückgezogen wurden, behalten ihre Gültigkeit nach dem Ende des auf der Bescheinigung angegebenen Zeitraums bis zu dem Datum, das in Absatz 3a des vorliegenden Artikels für die jeweilige Risikoklasse der Produkte festgelegt ist. Bescheinigungen, die von Benannten Stellen im Einklang mit diesen Richtlinien ab dem 25. Mai 2017 ausgestellt wurden, am 26. Mai 2021 noch gültig waren und vor dem 20. März 2023 abgelaufen sind, gelten nur dann bis zu den in Absatz 3a des vorliegenden Artikels festgelegten Zeitpunkten als gültig, wenn eine der folgenden Bedingungen erfüllt ist:
a) Vor Ablauf der Bescheinigung haben der Hersteller und eine Benannte Stelle eine schriftliche Vereinbarung gemäß Anhang VII Abschnitt 4.3 Unterabsatz 2 der vorliegenden Verordnung über die Konformitätsbewertung des Produkts, für das die abgelaufene Bescheinigung gilt, oder eines Produkts, das dazu bestimmt ist, dieses Produkt zu ersetzen, unterzeichnet;
b) eine zuständige Behörde eines Mitgliedstaats hat eine Ausnahme von dem anwendbaren Konformitätsbewertungsverfahren gemäß Artikel 59 Absatz 1 der vorliegenden Verordnung gewährt oder den Hersteller gemäß Artikel 97 Absatz 1 der vorliegenden Verordnung aufgefordert, das anzuwendende Konformitätsbewertungsverfahren durchzuführen
MDR Artikel 120 (2)
Interpretation dieses Textes
Falls der Hersteller ein Zertifikat hat, welches unter den Richtlinien vor dem 26.05.2017 ausgestellt wurde, bleibt dieses bis zum angegebenen Datum gültig. Ausgenommen hiervon sind Zertifikate gemäß Anhang IV der Richtlinien (Batch-Zertifizierung), die bis zum maximal 27.05.2022 gültig waren.
Wenn das Zertifikat der Benannten Stelle hingegen am oder nach dem 26.05.2017 ausgestellt wurde und am 26.05.2021 noch gültig war, behält es seine Gültigkeit bis zu den genannten Fristen (siehe Absatz 3(a)). Wenn dieses Zertifikat allerdings vor Inkrafttreten der Änderungsverordnung, d.h. vor dem 20. März 2023, abgelaufen ist, gilt die verlängerte Gültigkeit nur, falls:
- der Hersteller VOR Ablauf des Zertifikats einen Vertrag mit einer Benannten Stelle zur MDR-Zertifizierung abgeschlossen hat (gemäß Anhang VII Abschnitt 4.3 Unterabsatz 2 MDR) und dieser unterzeichnet wurde
- ODER dem Hersteller eine Sondergenehmigung durch die nationale Behörde (in Deutschland das BfArM) gemäß Artikel 59 Absatz 1 MDR erteilt wurde
- ODER eine weitere Inverkehrbringung durch die zuständige Behörde (in Deutschland sind das die Landesbehörden) auch ohne gültiges Zertifikat unter Fristsetzung gestattet wurde (Artikel 97 Absatz 1 MDR).
b) MDR Artikel 120 Absatz 3 (a) und (b)
Originaltext
(3) Abweichend von Artikel 5 und unter der Voraussetzung, dass die in Absatz 3c genannten Bedingungen erfüllt sind, dürfen die in den Absätzen 3a und 3b genannten Produkte bis zu den in diesen Absätzen genannten Zeitpunkten in Verkehr gebracht oder in Betrieb genommen werden.
(3a) Produkte, für die gemäß der Richtlinie 90/385/EWG oder der Richtlinie 93/42/EWG eine aufgrund von Absatz 2 des vorliegenden Artikels gültige Bescheinigung ausgestellt wurde, dürfen bis zu den folgenden Zeitpunkten in Verkehr gebracht oder in Betrieb genommen werden:
a) 31. Dezember 2027 für alle Produkte der Klasse III und für implantierbare Produkte der Klasse IIb, ausgenommen Nahtmaterial, Klammern, Zahnfüllungen, Zahnspangen, Zahnkronen, Schrauben, Keile, Zahn- bzw. Knochenplatten, Drähte, Stifte, Klemmen und Verbindungsstücke;
b) 31. Dezember 2028 für andere Produkte der Klasse IIb als die unter Buchstabe a des vorliegenden Absatzes genannten, für Produkte der Klasse IIa und für Produkte der Klasse I, die in sterilem Zustand in Verkehr gebracht werden, oder mit Messfunktion.
(3b) Produkte, für die das Konformitätsbewertungsverfahren gemäß der Richtlinie 93/42/EWG nicht die Mitwirkung einer Benannten Stelle erforderte, für die die Konformitätserklärung vor dem 26. Mai 2021 ausgestellt wurde und für die das Konformitätsbewertungsverfahren gemäß der vorliegenden Verordnung die Mitwirkung einer Benannten Stelle erfordert, dürfen bis zum 31. Dezember 2028 in Verkehr gebracht oder in Betrieb genommen werden.
MDR Artikel 120 (3)
Interpretation dieses Textes
Der dritte Absatz des Artikel 120 (a) und (b) lässt sich wie folgt deuten:
Legacy-Produkte mit gültigem Richtlinien-Zertifikat dürfen abhängig von der Risikoklasse bis zu den genannten Fristen in Verkehr gebracht werden.
- Klasse III, implantierbare Produkte der Klasse IIb (ausgenommen die Produkte mit „Well-Established-Technology“): 31.12.2027
- Sonstige Klasse IIb, IIa, Is und Im Produkte: 31.12.2028
- Produkte der Klasse I unter den Richtlinien, welche unter der MDR höher klassifiziert wurden und deshalb eine Benannte Stelle im Konformitätsbewertungsverfahren einbezogen werden muss (z. B. wiederverwendbare chirurgische Instrumente, viele Software-Produkte): 31.12.2028
Diese erweiterten Übergangsfristen gelten allerdings nur unter bestimmten Bedingungen. Diese sind in Absatz 3 (c) genannt.
c) MDR Artikel 120 Absatz 3 (c)–(d)
Originaltext
(3c) Produkte gemäß den Absätzen 3a und 3b des vorliegenden Artikels dürfen nur bis zu den in diesen Absätzen genannten Zeitpunkten in Verkehr gebracht oder in Betrieb genommen werden, wenn die folgenden Bedingungen erfüllt sind:
a) Die Produkte entsprechen weiterhin der Richtlinie 90/385/EWG bzw. der Richtlinie 93/42/EWG;
b) es liegen keine wesentlichen Veränderungen der Auslegung und der Zweckbestimmung vor;
c) die Produkte stellen kein unannehmbares Risiko für die Gesundheit oder Sicherheit der Patienten, Anwender oder anderer Personen oder für andere Aspekte des Schutzes der öffentlichen Gesundheit dar;
d) der Hersteller hat spätestens am 26. Mai 2024 ein Qualitätsmanagementsystem gemäß Artikel 10 Absatz 9 eingerichtet;
e) der Hersteller oder der Bevollmächtigte hat spätestens am 26. Mai 2024 bei einer Benannten Stelle einen förmlichen Antrag gemäß Anhang VII Abschnitt 4.3 Unterabsatz 1 auf Konformitätsbewertung für ein in dem Absatz 3a oder 3b genanntes Produkt oder für ein Produkt, das dazu bestimmt ist, dieses Produkt zu ersetzen, gestellt, und die Benannte Stelle und der Hersteller haben spätestens am 26. September 2024 eine schriftliche Vereinbarung gemäß Anhang VII Abschnitt 4.3 Unterabsatz 2 unterzeichnet.
(3d) Abweichend von Absatz 3 des vorliegenden Artikels gelten die Anforderungen dieser Verordnung an die Überwachung nach dem Inverkehrbringen, die Marktüberwachung, die Vigilanz, die Registrierung von Wirtschaftsakteuren und von Produkten für Produkte gemäß den Absätzen 3a und 3b anstelle der entsprechenden Anforderungen der Richtlinien 90/385/EWG und 93/42/EWG.
MDR Artikel 120 (3)
Interpretation dieses Textes
Der dritte Absatz des Artikel 120 (c) beschreibt die Bedingungen, welche für die Anwendung der erweiterten Übergangsfristen erfüllt sein müssen.
Voraussetzung 1
Das Produkt entspricht weiterhin den Anforderungen der MDD bzw. AIMDD. Das klingt logisch und sollte durch die Überwachung der Benannten Stellen und Behörden auch sichergestellt werden.
Voraussetzung 2
Der Hersteller hat keine wesentlichen Änderungen an dem Produkt oder an seiner Zweckbestimmung vorgenommen. Was man genau darunter versteht, erklärt das MDCG im Dokument MDCG 2020-03.
Besonderen Anlass zu Diskussionen bietet die Frage, wie groß Änderungen sein dürfen, damit sie nicht als „wesentlich“ betrachtet werden müssen. Beachten Sie hierzu die Artikel zu Design Changes und Software Changes.
Voraussetzung 3
Dass die Produkte kein unannehmbares Risiko für die Sicherheit und Gesundheit darstellen, sollte durch Einhaltung der Richtlinien sichergestellt sein. Dennoch ist dies eine Überwachungsaufgabe der Benannten Stellen und Überwachungsbehörden. Das stellt auch das MDCG im Guidance-Dokument MDCG 2022-04 klar. Dieses enthält Empfehlungen zu den Überwachungstätigkeiten von Legacy-Produkten während der Übergangsfristen. Es heißt dort:
„In cases where the audit activities reveal a major non-conformity, which may present an
unacceptable risk to the health or safety of patients, users or other persons, the notified body
needs to take action, i.e. suspend, restrict or withdraw the certificate, and inform the relevant
competent authority.“
Voraussetzung 4
Die Forderungen nach einem vollständigen QM-System gemäß Artikel 10 Absatz 9 wurde durch die Änderungsverordnung ergänzt. Somit reicht es nicht mehr aus, nur die Prozesse für die Überwachung nach Inverkehrbringen und Vigilanz gemäß MDR zu implementieren. Letztere müssen bereits seit dem 26.05.2021 MDR-konform implementiert sein (siehe 3(d)).
Voraussetzung 5
Auch diese Bedingung ist nicht zu unterschätzen und darf nicht vernachlässigt werden. Hersteller müssen bis zum 26.05.2024 einen Antrag auf MDR-Zertifizierung bei einer Benannten Stelle gestellt haben. Unklar ist, ob bis dahin der Antrag nur gestellt sein muss oder ob dieser durch die Benannte Stelle auch positiv geprüft worden sein muss. Dies lässt Raum für Interpretation und Meinungsverschiedenheiten. Wir gehen aktuell davon aus, dass dieses Datum allein die Antragstellung betrifft, aber nicht die Prüfung durch die Benannte Stelle. Interessant ist die Möglichkeit, die MDR-Zertifizierung nicht für das eigentliche Legacy-Produkt, sondern für ein „ersetzendes Produkt“ zu beantragen. Unklar ist dabei allerdings, welche Voraussetzungen dafür gelten. Muss das ersetzende Produkt äquivalent sein oder darf unterschiedliche Technologie eingesetzt werden? Auch dies lässt leider Raum für Interpretation. Darüber sollte die MDCG schnellstmöglich Klarheit schaffen.
Zusätzlich zur Antragsstellung muss bis zum 26.09.2024 ein unterschriebener Vertrag mit einer Benannten Stelle zur MDR-Zertifizierung vorliegen.
d) MDR Artikel 120 Absatz 3 (e)
Originaltext
(3e) Unbeschadet des Kapitels IV und des Absatzes 1 des vorliegenden Artikels bleibt die Benannte Stelle, die die Bescheinigung gemäß Absatz 3a des vorliegenden Artikels ausgestellt hat, für die angemessene Überwachung aller geltenden Anforderungen an die von ihr zertifizierten Produkte verantwortlich, es sei denn, der Hersteller ist mit einer Benannten Stelle, deren Benennung gemäß Artikel 42 erfolgt ist, übereingekommen, dass sie eine derartige Überwachung durchführt.
Spätestens am 26. September 2024 ist die Benannte Stelle, die die schriftliche Vereinbarung gemäß Absatz 3c Buchstabe e des vorliegenden Artikels unterzeichnet hat, für die Überwachung der unter die schriftliche Vereinbarung fallenden Produkte verantwortlich. Betrifft die schriftliche Vereinbarung ein Produkt, das dazu bestimmt ist, ein Produkt zu ersetzen, für das eine Bescheinigung gemäß der Richtlinie 90/385/EWG oder der Richtlinie 93/42/EWG ausgestellt wurde, so wird die Überwachung in Bezug auf das ersetzte Produkt durchgeführt.
Die Vorkehrungen für die Übertragung der Überwachung von der Benannten Stelle, die die Bescheinigung ausgestellt hat, auf die Benannte Stelle, deren Benennung gemäß Artikel 42 erfolgt ist, werden in einer Vereinbarung zwischen dem Hersteller und der Benannten Stelle, deren Benennung gemäß Artikel 42 erfolgt ist, und, soweit durchführbar, der Benannten Stelle, die die Bescheinigung ausgestellt hat, klar geregelt. Die Benannte Stelle, deren Benennung gemäß Artikel 42 erfolgt ist, ist nicht für Konformitätsbewertungstätigkeiten verantwortlich, die von der Benannten Stelle, die die Bescheinigung ausgestellt hat, durchgeführt werden.
MDR Artikel 120 (3)
Interpretation dieses Textes
Im Absatz 3(e) werden die Überwachungstätigkeiten und -zuständigkeiten der Benannten Stellen geregelt. Im Regelfall ist demnach die Benannte Stelle, die das Richtlinien-Zertifikat ausgestellt hat, für die weitere Überwachung innerhalb der Übergangsfristen zuständig. Allerdings dürfen Hersteller ihre Benannte Stelle wechseln, welche dann die Überwachungstätigkeiten übernehmen. Dies kann z. B. notwendig sein, wenn die aktuelle Benannte Stelle (noch) nicht unter der MDR benannt wurde. Spätestens zum 26.09.2024 muss dann eine Benannte Stelle, designiert unter der MDR, die Überwachungsverantwortung innehaben.
e) MDR Artikel 120 Absatz 3 (f)
Originaltext
(3f) Abweichend von Artikel 5 dürfen implantierbare Sonderanfertigungen der Klasse III bis zum 26. Mai 2026 ohne eine von einer Benannten Stelle nach dem Konformitätsbewertungsverfahren gemäß Artikel 52 Absatz 8 Unterabsatz 2 ausgestellte Bescheinigung in Verkehr gebracht oder in Betrieb genommen werden, sofern der Hersteller oder der Bevollmächtigte spätestens am 26. Mai 2024 bei einer Benannten Stelle einen förmlichen Antrag gemäß Anhang VII Abschnitt 4.3 Unterabsatz 1 auf Konformitätsbewertung gestellt hat und die Benannte Stelle und der Hersteller spätestens am 26. September 2024 eine schriftliche Vereinbarung gemäß Anhang VII Abschnitt 4.3 Unterabsatz 2 unterzeichnet haben.
MDR Artikel 120 (3)
Interpretation dieses Textes
Absatz 3(f) wurde mit der Änderungsverordnung ergänzt. Nun gewährt die MDR auch für implantierbare Sonderanfertigungen der Klasse III eine erweiterte Übergangsfrist bis zum 26.05.2026. Die Bedingungen sind in diesem Fall eingeschränkt. So verlangt die MDR „nur“ einen förmlichen Antrag zur Konformitätsbewertung bis zum 26.05.2024 und den unterzeichneten Vertrag bis zum 26.09.2024.
f) MDR Artikel 120 Absatz 4
Originaltext
Der vierte Absatz ist vergleichsweise gut verständlich:
(4) Produkte, die vor dem 26. Mai 2021 gemäß den Richtlinien 90/385/EWG und 93/42/EWG rechtmäßig in Verkehr gebracht wurden, und Produkte, die ab dem 26. Mai 2021 gemäß den Absätzen 3, 3a, 3b und 3f in Verkehr gebracht wurden, dürfen weiter auf dem Markt bereitgestellt oder in Betrieb genommen werden.
MDR Artikel 120(4)
Interpretation des Textes
Damit ist der Abverkauf zeitlich nicht mehr limitiert. Diese Regelung soll sicherstellen, dass „sichere und wichtige Medizinprodukte, die bereits in Verkehr gebracht wurden, den Gesundheitssystemen sowie den Patientinnen und Patienten, die darauf angewiesen sind, weiterhin zur Verfügung stehen.“
Zu unterscheiden sind hierbei die Begriffe „Bereitstellung“ und „Inverkehrbringung“. Eine Definition der Begriffe finden Sie in unserem Artikel zur Inverkehrbringung.
g) MDR Artikel 120 Absatz 8
Der achte Absatz erzeugt bei vielen Lesern nur Kopfschütteln. Wer soll das beim ersten Lesen verstehen?
„(8) Abweichend von Artikel 10a, Artikel 10b Absatz 1 Buchstabe a und Artikel 11 Absatz 5 der Richtlinie 90/385/EWG und von Artikel 14 Absätze 1 und 2, Artikel 14a Absatz 1 Buchstaben a und b und Artikel 16 Absatz 5 der Richtlinie 93/42/EWG wird angenommen, dass Hersteller, Bevollmächtigte, Importeure und Benannte Stellen, die im Zeitraum, der am späteren der in Artikel 123 Absatz 3 Buchstabe d genannten Daten beginnt und 18 Monate später endet, Artikel 29 Absatz 4, Artikel 31 Absatz 1 und Artikel 56 Absatz 5 der vorliegenden Verordnung genügen, die Vorschriften und Bestimmungen erfüllen, die die Mitgliedstaaten nach Maßgabe des Beschlusses 2010/227/EU gemäß Artikel 10a der Richtlinie 90/385/EWG bzw. gemäß Artikel 14 Absätze 1 und 2 der Richtlinie 93/42/EWG, gemäß Artikel 10b Absatz 1 Buchstabe a der Richtlinie 90/385/EWG bzw. gemäß Artikel 14a Absatz 1 Buchstaben a und b der Richtlinie 93/42/EWG sowie gemäß Artikel 11 Absatz 5 der Richtlinie 90/385/EWG bzw. gemäß Artikel 16 Absatz 5 der Richtlinie 93/42/EWG erlassen haben.“
MDR Artikel 120 (8)
Interpretation des Textes
Im Kontext der EUDAMED stellt die MDR die folgenden Anforderungen an Legacy-Produkte:
- Die Hersteller müssen Produkte (außer Sonderanfertigungen) in der EUDAMED registrieren (Artikel 29 Absatz 4).
- Die Wirtschaftsakteure müssen sich selbst in der EUDAMED registrieren (Artikel 31 Absatz 1).
- Die Benannten Stellen müssen die Bescheinigungen (Zertifikate von Benannten Stellen) in der EUDAMED hinterlegen (Artikel 56 Absatz 5).
Artikel 120 Absatz 8 sagt somit aus, dass etwa Hersteller vom Legacy-Produkten, die sich und ihre Produkte in der EUDAMED registrieren, den entsprechenden Verpflichtungen gemäß den alten Richtlinien nachkommen.
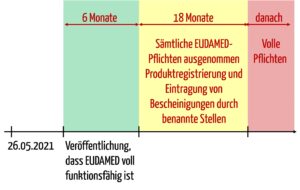
Allerdings bezieht sich diese Pflicht auf den „Zeitraum, der am späteren der in Artikel 123 Absatz 3 Buchstabe d genannten Daten beginnt und 18 Monate später endet“.
In diesem Buchstaben d sind die folgenden Daten zu erkennen:
- 26.05.2021 (Geltungsbeginn der MDR)
- Der Tag sechs Monate nach dem Tag der Veröffentlichung der Bekanntmachung gemäß Artikel 34 Absatz 3. In diesem Artikel geht es um die Veröffentlichung im Amtsblatt, die besagt, dass die EUDAMED voll funktionsfähig ist.
Das bedeutet, dass im Kontext von EUDAMED und Registrierung die Hersteller in einem Zeitraum von 18 Monaten „nur“ die o. g. Pflichten zu erfüllen haben. Danach gelten alle Anforderungen bezüglich der EUDAMED.
Für alle, die den Absatz 8 noch genauer verstehen wollen, hier wichtige Referenzen:
- AIMDD (90/385/EWG)
- Artikel 10a: Registrierung des Herstellers
- Artikel 10b Absatz 1 Buchstabe a: Bescheinigungen in Datenbank
- Artikel 11 Absatz 5: von Benannten Stellen ausgestellte und zurückgezogene Bescheinigungen
- MDD (93/42/EWG)
- Artikel 14 Absätze 1 und 2: Meldung der für das Inverkehrbringen verantwortlichen Personen
- Artikel 14a Absatz 1 Buchstaben a und b: Europäische Datenbank (Hersteller, Bevollmächtigte, Bescheinigungen)
- Artikel 16 Absatz 5: von Benannten Stellen ausgestellte und zurückgezogene Bescheinigungen
h) MDR Artikel 123 („Inkrafttreten und Geltungsbeginn“)
Der Artikel 123 nennt weitere Fristen, die die Wirtschaftsakteure betreffen, z. B.:
- Absatz 3 Buchstabe d: Im Kontext der EUDAMED sind hier alle Pflichten aufgelistet, die spätestens sechs Monate nach dem Zeitpunkt erfüllt werden müssen, an dem die EUDAMED als voll funktionsfähig erklärt wird. Im Gegensatz zu Artikel 120 Absatz 8 geht es hier nicht um Übergangsbestimmungen für alte Produkte.
- Absatz 3 Buchstabe e: Für die Registrierung von Produkten und die Hinterlegung von Bescheinigungen in der EUDAMED gewährt die MDR sogar 6 + 18 Monate Kulanz, beginnend mit dem Zeitpunkt, zu dem die EUDAMED als voll funktionsfähig erklärt wird.
- Der UDI-Träger muss bei implantierbaren Produkten sowie Produkten der Klasse III ab dem 26.05.2021 angebracht werden, bei Produkten der Klasse IIa und IIb ab dem 26.05.2023 und für Produkte der Klasse I ab dem 26.05.2025. Für die Vergabe der UDI gilt diese Ausnahmeregelung nicht.
4. Sonstige Anforderungen
Folgende Dokumente sind regulatorisch relevant und sollten beachtet werden:
5. Fazit und Zusammenfassung
Der Druck war groß, denn die Beschwerden über die negativen Auswirkungen der MDR konnte die EU-Kommission nicht ignorieren. Daher hat sie mehrfach die Übergangsfristen verschoben. Die Probleme sind dadurch aber nur teilweise behoben. Viele Schäden sind bereits eingetreten:
- Hersteller haben aufgegeben.
- Produkte wurden vom Markt genommen.
- Neue Produkte werden zuerst in den USA zugelassen.
- Die Entwicklung neuer Produkte wurde aufgegeben oder zurückgestellt.
Dennoch war die Verlängerung der Übergangsfristen ein wichtiger Schritt. Die mehrfach überarbeiteten Vorgaben sind allerdings inzwischen schwer zu überblicken und lassen noch Fragen offen.
Änderungshistorie
- 2024-06-07: Kapitel 3 c): Referenz und Zitat aus dem MDCG 2022-4 aktualisiert
- 2023-08-25: In Kapitel 2 a) Flowchart der EU ergänzt
- 2023-03-20: Datum des Inkrafttretens der Änderungsverordnung ergänzt
- 2023-02-24: Artikel komplett überarbeitet nach Verabschiedung durch Parlament
- 2023-02-16: Hinweis, dass Vorschlag dem Parlament vorliegt, ergänzt
- 2023-01-12: Ausblick sowie Link zur Kommentierungsmöglichkeit ergänzt
- 2023-01-10: Hinweis zu geplanten Änderungen der Übergangsfristen ergänzt
- 2022-03-04: Hinweis zu MDCG 2022-4 ergänzt
- 2021-06-14: Link zum überarbeiteten Artikel zur PRRC eingefügt, der die diesbezüglichen Übergangsfristen thematisiert
- 2021-05-17: In Kapitel 4d) die Hinweise zu den UDI-Anforderungen bei Legacy-Devices ergänzt

