
Der europäische Verordnung über Medizinprodukte MDR (EU-Medizinprodukteverordnung) müssen von Herstellern berücksichtigt werden, die Medizinprodukte in der EU vermarkten wollen.
Das Verordnung (EU) 2017/745 über MedizinprodukteDer offizielle Titel stellt auch Anforderungen an benannte Stellen, Händler, Importeure und Gesundheitseinrichtungen wie Krankenhäuser.
… für Anfänger
Wenn Sie ganz neu sind, laden Sie es herunter kostenloses Starter-Kit runter. Es gibt Ihnen einen Überblick über die Vorschriften, zeigt Ihnen die Schritte zur „Zulassung“ Ihres Medizinprodukts und enthält die MDR-Checkliste im PDF- und DOCX-Format zum Download.
… für fortgeschrittene Benutzer
Erfahren Sie mehr über die Details auf dieser Seite und den darin verlinkten Fachartikeln.
Benutze das Konsolidierte Version der MDR in Deutsch oder Englisch. Hier sind alle Änderungen der MDR inklusive der im März 2023 verlängerten Übergangsfristen zusammengefasst. Interne Links erleichtern Ihnen die Orientierung in der über 170 Seiten umfassenden Verordnung.
… für IVDR-Hersteller
Hersteller von In-vitro-Diagnostika sollten den Fachartikel zu IVDR lesen.
… für Importeure, Händler und Betreiber
Schauen Sie sich das folgende Video an und lesen Sie anschließend die in Kapitel 1.b) verlinkten Fachartikel.
1. Überblick über die Anforderungen der MDR
1.1 Anforderungen an Hersteller und deren Medizinprodukte
Die Anforderungen, die die Medizinprodukteverordnung an Hersteller stellt, sind umfangreich.
1.1.1 Produktübergreifende Anforderungen
| MDR-Anforderung und Links zu Fachartikeln |
Erläuterung |
| Qualitätsmanagementsystem | Alle Hersteller benötigen ein QM-System unter anderem für die Entwicklung, Produktion und Überwachung der Produkte auf dem Markt. Mit Ausnahme von Produkten der Klasse I ist in der Regel eine Zertifizierung erforderlich. |
| Risikomanagementsystem | Das Risikomanagement muss während des gesamten Produktlebenszyklus sicherstellen, dass der Nutzen angesichts der Risiken akzeptabel ist. |
| Verantwortliche Person („Person, die für die Einhaltung gesetzlicher Vorschriften verantwortlich ist“) | Hersteller sind verpflichtet, eine Person zu beschäftigen, die für die Einhaltung gesetzlicher Vorschriften verantwortlich ist und z. B. dafür sorgt, dass die technische Dokumentation gemäß dem unternehmenseigenen QM-System erstellt wird. |
1.1.2 Produktspezifische Anforderungen
| MDR-Anforderung und Links zu Fachartikeln |
Erläuterung |
| Klassifizierung von Produkten | Der Hersteller muss für jedes Produkt die Risikoklasse ermitteln. |
| Grundlegende Sicherheits- und Leistungsanforderungen und technische Dokumentation | Die technische Dokumentation muss den Anforderungen des Anhangs II genügen und alle Nachweise darüber erbringen, dass die wesentlichen Anforderungen des Anhangs I erfüllt sind. Für diesen Nachweis sind die Hersteller verpflichtet, harmonisierte Normen und die gemeinsamen Spezifikationen zu verwenden. |
| Klinische Bewertung | Im Rahmen der klinischen Bewertung muss der Hersteller kontinuierlich prüfen, ob Sicherheit, Leistung und Nutzen der Produkte erfüllt sind. Reichen die klinischen Daten nicht aus, ist eine klinische Studie erforderlich. |
| Eindeutige Geräteidentifikation (UDI) | Alle Medizinprodukte müssen eine eindeutige Identifikation, die UDI, erhalten. Die Produkte müssen daher in EUDAMED registriert werden. |
| Beschriftung | Die MDR legt Anforderungen an die Gebrauchsanweisung, weitere Begleitmaterialien und sonstige Kennzeichnungen wie Aufdrucke und Verpackungen genau fest. |
| Konformitätsbewertung | Abhängig von der Risikoklasse des Produkts kann der Hersteller ein Konformitätsbewertungsverfahren durchlaufen. Dabei muss er – außer bei Produkten der Klasse I – eine benannte Stelle einschalten. Im Erfolgsfall muss er eine Konformitätserklärung ausfüllen und das CE-Zeichen anbringen. |
| Überwachung und Wachsamkeit nach dem Inverkehrbringen | Hersteller sind verpflichtet, ihre auf dem Markt befindlichen Medizinprodukte über deren gesamte Lebensdauer zu überwachen, kontinuierlich Daten zu sammeln und gegebenenfalls darauf zu reagieren. Wenn Risiken bestehen, müssen Sie die Behörden informieren. |
1.2 Anforderungen an andere Akteure (Händler, Importeure etc.)
Nicht nur Hersteller von Medizinprodukten müssen die Medizinprodukteverordnung einhalten, sondern auch andere Akteure:
2. Struktur und Aufbau der MDR
Die Medizinprodukteverordnung umfasst 123 Artikel, die in 10 Kapitel unterteilt sind. Es enthält außerdem 17 Anhänge.
2.1 Die Kapitel
Die Medical Device Regulation (MDR) ist im Vergleich zur alten Medical Device Directive (MDD) komplett neu strukturiert:
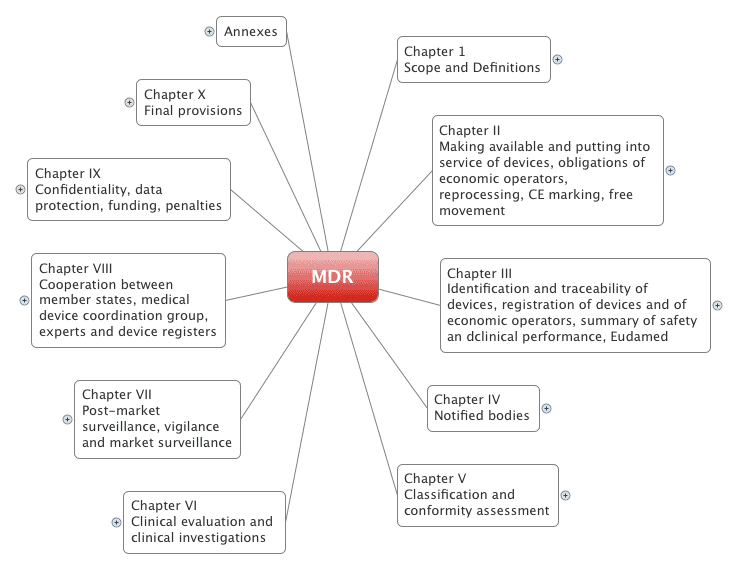
Es bietet einen schnellen Überblick über die MDR, die mehr als 170 Seiten umfasst.
2.2 Die Anhänge
Die MDR hat 17 Anhänge.
3. Weitere Fachartikel und Links
3.1 Weitere Fachartikel
Die EU hat die Übergangsfristen für die Umstellung auf die MDR mehrfach verschoben. Die Regelungen sind so komplex, dass ein Fachartikel zu den Übergangsfristen weiterhelfen kann.
Die Medical Device Coordination Group (MDCG) hat weitere Erläuterungen und Anforderungen veröffentlicht.
Für Produkte ohne medizinischen Zweck („Annex XVI-Produkte“) gelten unterschiedliche Anforderungen.
3.2 Weiterführende Links
- MDR (Originaltext)
- Zu korrigieren
- Verschiedenes
4. Hintergrundinformationen
4.1 Wie es zu den EU-Verordnungen kam
Oft wird gemunkelt, dass der Brustimplantat-Skandal der Auslöser für die Revision des Medizinproduktegesetzes gewesen sei. Doch die meisten Schauspieler bestreiten das mittlerweile. Es ist daher weitgehend unklar, wer die Neuregelung aus welchem Grund initiiert hat.
4.2 Unterschied zwischen EU-Verordnungen und EU-Richtlinien
Die „alten“ EU-MedizinprodukteRichtlinien Wie alle EU-Richtlinien mussten sie in nationale Gesetze und nationale Verordnungen umgesetzt werden, um Rechtskraft zu erlangen. In Deutschland waren dies das Medizinproduktegesetz (MPG) und Verordnungen wie die Medizinprodukte-Betreiberverordnung (noch gültig) und die Medizinprodukte-Sicherheitsplan-Verordnung (inzwischen ungültig).
Die EU-Vorschriften (hier: MDR, IVDR) haben unmittelbaren Rechtscharakter. Nationale Gesetze ergänzen diese Regelungen lediglich, wie unter anderem das MPDG in Deutschland, durch strafrechtliche Bestimmungen und die Festlegung der zuständigen Behörden.
4.3 Kritik an der Medizinprodukteverordnung
Die Kritik an der EU-Medizinprodukteverordnung ist massiv. Aufwand und Kosten für die Hersteller haben sich vervielfacht. Es gilt auch die Dauer des „Genehmigungsverfahrens“.
Dadurch werden Wettbewerbsfähigkeit, Innovation und die Versorgung mit Medizinprodukten behindert.
Diese Folgen waren bereits im Jahr 2020 zu befürchten.

Mit dem Laden des Videos akzeptieren Sie die Datenschutzbestimmungen von YouTube.
Erfahren Sie mehr
Video laden
==
Für die zukünftige Regulierung ist es wichtig, das System zu verstehen – eine Aufgabe der Regulierungswissenschaft.
5. Fazit und Zusammenfassung
Bei der Medizinprodukteverordnung MDR handelt es sich um ein sehr umfangreiches Gesetz, das alle Akteure (Medizinproduktehersteller, Benannte Stellen, Importeure, Händler, Krankenhäuser) vor große Herausforderungen stellt. Es ist nicht ersichtlich, dass die MDR die Sicherheit, Leistung und Wirksamkeit von Medizinprodukten verbessert. Es ist jedoch klar, dass die Versorgung mit Medizinprodukten leidet.
Den Herstellern bleibt nichts anderes übrig, als sich intensiv mit diesem Gesetzeswerk auseinanderzusetzen und seinen Anforderungen gerecht zu werden. Diese erhöhten Anforderungen beschränken sich nicht nur auf die „Zulassung“ (Pre-Market-Phase), sondern betreffen explizit auch die Post-Market-Phase (Post-Market-Überwachung, Vigilanz).
Hilfe bei der Umsetzung der MDR
Kostenlose Angebote
Sie haben noch Fragen zur MDR und ihrer Umsetzung? Antworten erhalten Sie in unserer kostenlosen Mikroberatung.
Laden Sie dies herunter kostenloses Starter-Kit Das gibt Ihnen einen Überblick über die Regulierungslandschaft und enthält die MDR-Checkliste im PDF- und DOCX-Format.
Videos und E-Learning
Die Videoschulungen in Auditgarant zeigen Ihnen Schritt für Schritt, wie Sie Ihre technische Dokumentation und Ihr QM-System schlank, schnell und MDR-konform erstellen. Es stehen über 100 Vorlagen und Beispieldokumente zum Download bereit. Damit schaffen Sie die Voraussetzungen dafür, dass Ihre Produkte schnell und sicher zugelassen und auf den Markt gebracht werden.
Produkttests
Die Experten des Johner Instituts helfen Ihnen beim Testen Ihrer Produkte:
Beratung
Nutzen Sie die Expertise der Regulatory Affairs-Experten des Johner Instituts, um…
Nehmen Sie jetzt Kontakt auf, damit wir gemeinsam klären können, wie Sie schnell und einfach die regulatorischen Anforderungen der MDR erfüllen und Ihre Produkte sicher auf den Markt bringen können.
Kontakt aufnehmen
Änderungshistorie (ab Mai 2021)
- 27.08.2024: In Kapitel 3.2 wurden die Links zu den Korrigenda korrigiert und die Links hierarchisch gruppiert
- 18.01.2024: Kapitelnummerierung geändert
- 18.04.2023: Artikel komplett neu geschrieben
- 10.10.2021: Abschnitt 7.e) (Die Konsequenzen wurden nicht berücksichtigt und werden nicht verstanden) hinzugefügt
- 26.07.2021: Link zum neuen Korrigendum hinzugefügt
- 24.05.2021: Neuer Beitrag zur Konformitätserklärung verlinkt


