Die Medizinprodukte-Betreiberverordnung (MPBetreibV) ist eine nationale deutsche Verordnung, die Anforderungen an das Erstellen, Betreiben und Anwenden von Medizinprodukten stellt.
1. FAQ zur MPBetreibV
a) Für wen gilt die Betreiberverordnung?
Die Medizinprodukte-Betreiberverordnung gilt für Organisation und Personen, die Medizinprodukte und In-vitro-Diagnostika anwenden und betreiben.
Typische Organisationen sind Krankenhäuser, Kliniken, Arzt-Praxen, Reha-Einrichtungen und medizinische Labore.
Organisationen und Personen, die Medizinprodukte errichten, bereithalten, instand halten, aufbereiten oder sicherheits- und messtechnischen Kontrollen unterziehen fallen ebenfalls in den Anwendungsbereich der Verordnung. Mehr dazu in Kapitel 3 dieses Artikels.
b) Welches Ziel verfolgt die MPBetreibV?
Ausgestaltung des Gesetzes
Formal verfolgt der Gesetzgeber mit der MPBetreibV das Ziel, die gesetzlichen Anforderungen des Medizinprodukterecht-Durchführungsgesetzes MPDG noch genauer zu formulieren.
Beispielsweise schreibt das MPDG:
Produkte […] dürfen nur nach Maßgabe der Rechtsverordnung nach § 88 Absatz 1 Satz 1 Nummer 6 betrieben und angewendet werden.
MPDG § 11
Was beim Betrieb dieser Produkte beachtet werden muss, sagt das MPDG nicht, sondern verweist auf die Rechtsverordnung – die Medizinprodukte-Betreiberverordnung.
Sicherheit von Patienten
Als Teil des Rechtssystems bestehend aus EU-Verordnungen (MDR, IVDR), Gesetzen (MPDG) und anderen nationalen Verordnungen verfolgt die MPBetreibV auch die Ziele dieses Rechtssystems: die Sicherheit, Leistungsfähigkeit und Wirksamkeit und damit auch die Sicherheit der Patienten sicherzustellen.
c) Was fordert die MPBetreibV?
Die Anforderungen beschreibt das zweite Kapitel dieses Artikels.
d) Wer ist für die Einhaltung der MPBetreibV verantwortlich?
Dass sich die MPBetreibV an die oben genannten Betreiber und Anwender richtet, sind diese für die Einhaltung dieser Rechtsvorschrift verantwortlich.
Die Überprüfung, dass dies tatsächlich geschieht, liegt in der Verantwortung der Bundesländer und ihrer Behörden wie ihren Regierungspräsidien und Gewerbeaufsichtsämtern.
e) Was muss man ins Bestandsverzeichnis eintragen?
Gesetz und Definition
Die MPBetreibV schreibt dazu:
Der Betreiber hat für alle aktiven nichtimplantierbaren Medizinprodukte der jeweiligen Betriebsstätte ein Bestandsverzeichnis zu führen. Die Aufnahme in ein Verzeichnis, das auf Grund anderer Vorschriften geführt wird, ist zulässig.
§ 13 MPBetreibV
Was aktive Medizinprodukte sind, definiert die MDR:
Ein Produkt, dessen Betrieb von einer Energiequelle mit Ausnahme der für diesen Zweck durch den menschlichen Körper oder durch die Schwerkraft erzeugten Energie abhängig ist und das mittels Änderung der Dichte oder Umwandlung dieser Energie wirkt. Ein Produkt, das zur Übertragung von Energie, Stoffen oder anderen Elementen zwischen einem aktiven Produkt und dem Patienten eingesetzt wird, ohne dass dabei eine wesentliche Veränderung von Energie, Stoffen oder Parametern eintritt, gilt nicht als aktives Produkt.
Software gilt ebenfalls als aktives Produkt;
Artikel 2, Absatz 4 MDR
Beispiele für Produkte, die im Bestandsverzeichnis geführt werden müssen
Medizinprodukte, die in das Bestandsverzeichnis aufgenommen werden müssen, sind:
- Medizinisch-elektrische Geräte wie Röntgengeräte, CTs, Ultraschallgeräte, Patientenmonitore, elektrische Blutdruckmessgeräte, Defibrillatoren
- Software als Medizinprodukt, beispielsweise PACS, die meisten Radiologie-Informations-Managementsysteme (RIS) und Patientendaten-Managementsysteme (PDMS), sofern es sich um Medizinprodukte handelt
- Elektrisch betriebene Rollstühle
- Aktive Produkte, die speziell für die Reinigung, Desinfektion oder Sterilisation von Medizinprodukten vorgesehen sind
- Laborgeräte, weil diese eine Untermenge der Medizinprodukte bilden
Streng genommen müssten sogar elektrische Fieberthermometer aufgenommen werden.
Beispiele für Produkte, die NICHT im Bestandsverzeichnis geführt werden müssen
Hingegen unterliegen diese Produkte nicht der Pflicht, in ein Bestandsverzeichnis aufgenommen zu werden:
- Chirurgische Instrumente
- Blutdruckmessgeräte und Stethoskope, die ohne Strom genutzt werden
- Krankenhaus-Informationssysteme (wenn diese kein Medizinprodukt sind)
- Rollstühle, die ohne Strom betrieben werden
2. MPBetreibV: Die wichtigsten Anforderungen
a) Anforderungen an das Betreiben
Die Medizinprodukte-Betreiberverordnung stellt Anforderungen an das Betreiben. Darunter zählt sie u. a.:
- Instandhaltung von Medizinprodukten, einschließlich der Prüfung „der für die Sicherheit und Funktionstüchtigkeit der Medizinprodukte wesentlichen konstruktiven und funktionellen Merkmale“ (§ 7 MPBetreibV)
- Aufbereitung der Medizinprodukte (§ 8 MPBetreibV)
- Einweisung der Anwender (§ 4 und § 10 MPBetreibV)
- Spätestens alle zwei Jahre die Durchführung von sicherheitstechnischen Kontrollen STK (§ 11 MPBetreibV) (s. u.)
- Führen eines Medizinproduktebuchs (§ 12 MPBetreibV) und Bestandsverzeichnisses (§ 13 MPBetreibV)
- Die Durchführung von messtechnischen Kontrollen MTK (§ 14 MPBetreibV) (s. u.)
Ein weiterer Artikel zur Instandhaltung gibt konkrete Hinweise, was bei der Wartung und Instandhaltung von Medizinprodukten zu beachten ist.
b) Anforderungen an den Betreiber und an Personen
Es gibt Anforderungen an den Betreiber bzw. die Personen selbst, z. B. in § 4 MPBetreibV:
(2) Medizinprodukte dürfen nur von Personen betrieben oder angewendet werden, die die dafür erforderliche Ausbildung oder Kenntnis und Erfahrung besitzen.
§ 4 MPBetreibV
Diese Forderungen richten sich nicht nur an Ärzte. Ärzte verfügen auch nicht notwendigerweise über alle Kompetenzen, um Medizinprodukte zu betreiben. Beispielsweise sind bei Software auch Tätigkeiten notwendig wie das Update des Betriebssystems, der Laufzeitumgebung, das Aktualisieren von Firewalls und Antivirus-Programmen, um den sicheren Betrieb sicherzustellen.
c) Anforderungen an das Risikomanagement
Die Sicherheit von Patienten, Anwendern und Dritten steht auch bei der MPBetreibV im Mittelpunkt. Sie fordert:
Der Betreiber hat die ihm nach dieser Verordnung obliegenden Pflichten wahrzunehmen, um ein sicheres und ordnungsgemäßes Anwenden der in seiner Gesundheitseinrichtung am Patienten eingesetzten Medizinprodukte zu gewährleisten.
MPBetreibV, § 3 (1)
Die gilt explizit auch für vernetzte Medizinprodukte:
Miteinander verbundene Medizinprodukte sowie mit Zubehör einschließlich Software oder mit anderen Gegenständen verbundene Medizinprodukte dürfen nur betrieben und angewendet werden, wenn sie zur Anwendung in dieser Kombination unter Berücksichtigung der Zweckbestimmung und der Sicherheit der Patienten, Anwender, Beschäftigten oder Dritten geeignet sind.
MPBetreibV, § 4 (4)
Betreiber dieser „verbundenen Medizinprodukte sowie Zubehör einschließlich Software“ sollten weitere spezielle Regelungen beachten, um die Konformität mit den Anforderungen der MPBetreibV zu gewährleisten. Beispielsweise wendet sich die Norm DIN EN IEC 80001-1 ebenfalls an die Betreiber. Sie stellt Anforderungen daran, wie diese das Risikomanagement für ihre medizinischen „IT-Netzwerke“ anwenden sollen.
Die IEC 80001-1 ist zwar gesetzlich nicht gefordert, jedoch schreibt der branchenspezifische Sicherheitsstandard (B3S) der Deutschen Krankenhausgesellschaft (DKG), dass die Anforderungen der DIN EN 80001-1 für den Einsatz von Medizingeräten in medizinischen IT-Netzwerken berücksichtigt werden sollen.
Exkurs: Seit 2022 sind alle Krankenhäuser in Deutschland durch § 75c, mittlerweile § 391des SGB V verpflichtet, nach dem Stand der Technik angemessene organisatorische und technische Vorkehrungen zur Sicherstellung der IT-Sicherheit ihrer informationstechnischen Systeme, Komponenten oder Prozesse zu treffen. Die Krankenhäuser können diese Verpflichtung erfüllen, wenn sie einen branchenspezifischen Sicherheitsstandard für die informationstechnische Sicherheit der Gesundheitsversorgung im Krankenhaus in der jeweils gültigen Fassung anwenden.
3. Betreiber im Sinne der Medizinprodukte-Betreiberverordnung (MPBetreibV)
a) Fehlende Definition des Begriffs „Betreiber“ vor 2017
Es gab lange keine Definition des Begriffs Betreiber, weder im MPG noch in der MPBetreibV. Daher mussten sich schon Gerichte damit beschäftigen herauszufinden, was der Gesetzgeber unter einem Betreiber versteht.
Urteil des Bundesverwaltungsgerichts
Hier ein Auszug aus einem Urteil:
Die Auslegung des Begriffs „Betreiber“ muss vom Wortsinn ausgehen. Das Wort „Betreiben“ wird in vielfältigen Zusammenhängen verwendet. Man kann zum Beispiel Studien, ein Geschäft, ein Handwerk, eine Angelegenheit, Ackerbau, Handel oder auch eine Liebhaberei betreiben (vgl. Wahrig, Deutsches Wörterbuch, Aufl. 2000, Stichwort „Betreiben“; Duden, Deutsches Universalwörterbuch, 2. Aufl., Stichwort „Betreiben“). Allen diesen Verwendungen ist gemeinsam, dass sie eine Tätigkeit bezeichnen. Insbesondere wenn vom Betreiben einer Maschine die Rede ist, steht für das allgemeine Sprachempfinden außer Frage, dass der tatsächliche Gebrauch und die Benutzung der Maschine gemeint sind. Betreiber ist danach derjenige, der – selbst oder durch seine Mitarbeiter – die Arbeit der Maschine steuert, sie an- und auch wieder abstellt und sie während des Betriebes überwacht. Entscheidend ist hierfür das Vorhandensein der tatsächlichen Sachherrschaft. An diesem Wortsinn orientiert sich ersichtlich auch die aufgeführte neuere Kommentarliteratur. Geht man davon aus, so liegt auf der Hand, dass der Verleiher eines Geräts, der dieses einem anderen zum Gebrauch und zur Nutzung überlässt, nicht Betreiber ist.
Das Fehlen einer gesetzlichen Definition des Begriffs Betreiber kann angesichts der Vielzahl anderer im Medizinproduktegesetz enthaltenen Definitionen nur dahin gedeutet werden, dass der Gesetzgeber diesen Begriff für eindeutig hielt und ihn im Sinne des allgemeinen Sprachgebrauchs verwendet wissen wollte.
b) Definition des Begriffs „Betreiber“ ab 2017
In der Novelle der MPBetreibV, die zum 01.01.2017 in Kraft trat, hat der Gesetzgeber nun endlich die fehlende Begriffsdefinition ergänzt:
Betreiber eines Medizinproduktes ist jede natürliche oder juristische Person, die für den Betrieb der Gesundheitseinrichtung verantwortlich ist, in der das Medizinprodukt durch dessen Beschäftigte betrieben oder angewendet wird. Abweichend von Satz 1 ist Betreiber eines Medizinproduktes, das im Besitz eines Angehörigen der Heilberufe oder des Heilgewerbes ist und von diesem zur Verwendung in eine Gesundheitseinrichtung mitgebracht wird, der betreffende Angehörige des Heilberufs oder des Heilgewerbes. Als Betreiber gilt auch, wer außerhalb von Gesundheitseinrichtungen in seinem Betrieb oder seiner Einrichtung oder im öffentlichen Raum Medizinprodukte zur Anwendung bereithält.
§ 2 MPBetreibV
Unter Betreiber versteht man intuitiv eher Institutionen als Personen. Typische Betreiber sind Krankenhäuser, Arztpraxen und medizinische Versorgungszentren. Abweichend davon gelten auch Angehörige der Heilberufe oder des Heilgewerbes, wie Belegärzte, als Betreiber, wenn sie ein Medizinprodukt zur Verwendung in einer Gesundheitseinrichtung mitbringen. Ebenso gilt als Betreiber, wer Medizinprodukte in seinem Betrieb, seiner Einrichtung oder im öffentlichen Raum zur Anwendung bereithält wie etwa ein Defibrillator in öffentlichen Einrichtungen. Der Betreiberbegriff im Medizinproduktrecht bezieht sich somit auf den Betrieb einer Gesundheitseinrichtung oder einer ausgelagerten „Gesundheitseinheit“ in Betrieben, Einrichtungen oder dem öffentlichen Raum.
Hingegen handelt es sich bei einem SaaS (Software as a Service) – Provider einer Medical Device Software (MDSW) nicht um einen Betreiber im Sinne der MPBetreibV, da die MDSW weder auf dessen unternehmensseitigem Server („in seinem Betrieb oder seiner Einrichtung“) zur Anwendung bereitgehalten, noch in einem sonstigen oder öffentlichen Betrieb bereitgestellt wird. Die MDSW wird stattdessen in einem begrenzten Kreis von (Gesundheits-)Einrichtungen auf Basis vertraglicher Vereinbarungen zur Anwendung zugänglich gemacht, wobei die Entscheidung über den Einsatz der MDSW vollständig beim Kunden verbleibt.
Wenn von „Betreiber“ bzw. „betreiben“ im SaaS-Kontext gesprochen wird, bezieht sich ausschließlich auf den technischen Betrieb der Anwendung. Der SaaS-Provider ist aber weder für den medizinischen Kontext, in dem seine MDSW eingesetzt wird, noch für die Umstände dieses Einsatzes verantwortlich und daher nicht mit dem Betreiber im Sinne der MPBetreibV gleichzusetzen. Er ist vergleichbar mit einem Dienstleister, der den technischen Betrieb von medizinischen Geräten in einer Klinik übernimmt. Auch dieser Dienstleister wird durch die Übernahme von Wartung, die den technischen Betrieb sicherstellt, nicht zum medizinprodukterechtlichen Betreiber.
Weil die MPBetreibV auch für Anwender relevant ist, hier noch die Definition dieses Begriffs:
Anwender ist, wer ein Medizinprodukt im Anwendungsbereich dieser Verordnung am Patienten einsetzt.
§ 2 MPBetreibV
4. MDR, MPDG und MPBetreibV
Die MDR gibt in Europa den rechtlichen Rahmen für Medizinproduktehersteller. Sie stellt auch einige Anforderungen an die Gesundheitseinrichtungen, z. B. bezüglich:
- Eigenherstellung
- Wiederaufbereitung
- UDI
Die MDR regelt aber nur in sehr eingeschränktem Umfang die Anwendung und den Betrieb der Medizinprodukte. Dies ist Aufgabe der MPBetreibV.
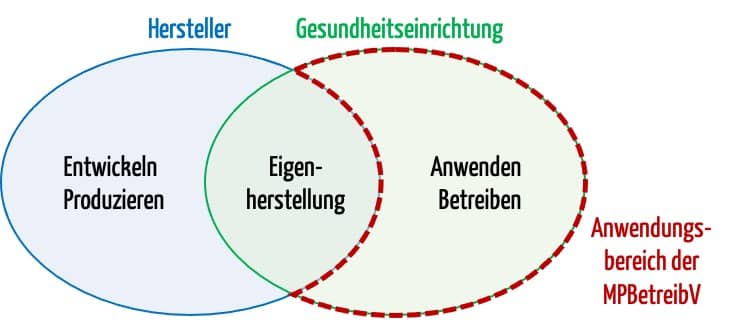
Nationale Verordnungen bedürfen immer eines Gesetzes, das eine entsprechende „Verordnungsermächtigung“ enthält. Im Falle der MPBetreibV ist das der § 88 im MPDG.
5. Sicherheitstechnische Kontrollen (STK) und messtechnische Kontrollen (MTK)
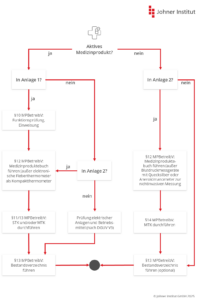
a) Sicherheitstechnische Kontrollen (STK)
Sicherheitstechnische Kontrollen (STK) haben das Ziel, die Sicherheit von Medizinprodukten zu überprüfen und sicherzustellen, um Gefährdungen für Patienten, Anwender und Dritte zu minimieren.
Die STK sind aber nur für die in Anlage 1 gelisteten Medizinprodukte vorgeschrieben. Das sind im Wesentlichen kritische aktive, nicht implantierbare Produkte wie Kernspingeräte, Stimulatoren, Beatmungsgeräte oder Geräte zur Lithotripsie.
Manche Hersteller verlangen in der Gebrauchsanweisung ihrer Medizinprodukte die Durchführung sicherheitstechnischer Kontrollen – auch für Produkte, die nicht in Anlage 1 aufgeführt sind. Unter dem Begriff „sicherheitstechnische Kontrolle“ verstehen sie dabei Prüfungen, die im Rahmen der Instandhaltung (§ 7 MPBetreibV) durchzuführen sind. Solche Kontrollen sind zu beachten. Sie sind jedoch strikt von den sicherheitstechnischen Kontrollen nach § 11 zu unterscheiden, die vom Verordnungsgeber vorgeschrieben sind.
Die MPBetreibV spricht in § 7 von Inspektionen und Wartungen:
„Inspektionen und Wartungen, die erforderlich sind, um den sicheren und ordnungsgemäßen Betrieb der Medizinprodukte fortwährend zu gewährleisten. Die Instandhaltungsmaßnahmen sind unter Berücksichtigung der Angaben des Herstellers durchzuführen, der diese Angaben dem Medizinprodukt beizufügen hat.“
§ 7 MPBetreibV
Umgekehrt können Hersteller die Betreiber nicht von den Pflichten zu STK befreien. Auch von der Gefährdungsbeurteilung und sicherheitstechnischen Bewertung (TRBS 1111) können die Hersteller die Betreiber nicht Fall entbinden. Vielmehr sollten die Betreiber die Angaben der Hersteller für diese Beurteilung nutzen.
Sicherheitstechnische Kontrollen sind spätestens alle zwei Jahre mit Ablauf des Monats durchzuführen, in dem die Inbetriebnahme des Medizinproduktes erfolgte oder die letzte sicherheitstechnische Kontrolle durchgeführt wurde. STK sind von qualifizierten Personen durchzuführen und zu dokumentieren.
b) Messtechnische Kontrollen (MTK)
Messtechnischen Kontrollen (MTK) haben das Ziel, die Messgenauigkeit von Medizinprodukten sicherzustellen. Die MPBetreibV nennt (in Anlage 2) die Produkte, die einer MTK unterzogen werden müssen. Dazu zählen unter anderem medizinische Elektrothermometer, nichtinvasive Blutdruckmessgeräte, Tretkurbelergometer und verschiedene Dosimeter.
Im Gegensatz zur STK, die spätestens alle zwei Jahre durchgeführt werden muss, gibt es bei der MTK unterschiedliche Nachprüffristen, die in Anlage 2 je Art des Medizinprodukts festgelegt sind. Dabei beginnen die Fristen mit Ablauf des Jahres, in dem das Medizinprodukt in Betrieb genommen oder die letzte MTK durchgeführt wurde.
Auch die MTK sind von qualifizierten Personen durchzuführen und zu dokumentieren.
Hersteller dürfen den Betreibern kürzere, aber keine längeren Prüffristen vorschreiben. Sie können die Betreiber nicht von der Pflicht zu MTK befreien.
7. Änderungen an der MPBetreibV
a) Änderungen zum 01.01.2017
Die Medizinprodukte-Betreiberverordnung (MPBetreibV) wurde im September 2016 überarbeitet. Die neue Version trat am 01.01.2017 in Kraft. Zu den Änderungen der Verordnung zählen:
- Die Verordnung enthält nun die Definition des Begriffs Betreiber.
- Anwender müssen sich nicht nur vom Funktionieren von Medizinprodukten überzeugen, sondern auch von dem Funktionieren verbundener Produkte. Der zunehmenden Vernetzung soll hier Rechnung getragen werden. Es bleibt unklar, wie Anwender diese Forderung erfüllen sollen.
- Gesundheitseinrichtungen mit mehr als 20 Mitarbeitenden müssen einen zentralen Ansprechpartner benennen und bereitstellen, den Beauftragten für Medizinproduktesicherheit. Die Anforderungen sind niedriger als jene an den Sicherheitsbeauftragten bei Herstellern.
- Eine Einweisung wird für alle Medizinprodukte gefordert. Ein bloßes „learning on the job“ ist nicht gestattet.
- Die sicherheitstechnischen Kontrollen sind nur noch für Produkte gefordert, die in Anlage 1 gelistet sind, es sei denn, der Hersteller fordert diese (mehr dazu weiter oben).
- Die messtechnischen Kontrollen sind nur noch für Produkte gefordert, die in Anlage 2 gelistet sind.
b) Änderungen im Jahr 2020/2021
Die Medizinprodukte-EU-Anpassungsverordnung (MPEUAnpV) hat die Medizinprodukte-Betreiberverordnung (MPBetreibV) geändert.
Die MPBetreibV wurde u. a. in den folgenden Punkten geändert werden:
- Anwendungsbereich: jetzt (auch) MDR und IVDR
- Verantwortlichkeiten der Behörde
- Regelungen für Einmalprodukte und deren Aufbereitung
- Referenzen
- Pflichten im Kontext der Implantationsausweise
Viele Änderungen sind formaler Natur. So mussten die Verweise auf die EU-Richtlinien in Verweise auf die EU-Verordnungen geändert werden.
Sie finden hier eine Gegenüberstellung der alten und neuen MPBetreibV.
c) Geplante Änderungen
Das Bundesministerium für Gesundheit (BMG) plant Änderungen an der MPBetreibV, welche der Referentenentwurf der dritten Verordnung zur Änderung medizinprodukterechtlicher Vorschriften aus November 2023 vorstellt.
Anlass der Änderungen
Hauptanlass vieler Änderungen ist die zunehmende Digitalisierung im Gesundheitswesen und der damit einhergehende vermehrte Einsatz von Medizinprodukte-Software, der Sorgen zu bereiten scheint, wie die Erwägungsgründe im Entwurf erkennen lassen:
Deutlich wurde, dass die zunehmende Digitalisierung neuer, auf ihre Besonderheiten zugeschnittener Regelungen bedarf. Es ist zu beobachten, dass Gesundheitseinrichtungen auch vermehrt auf den Einsatz von Medizinprodukte- Software bei den Behandlungen von Patienten zurückgreifen. Dies birgt verschiedene Risiken für das sichere Funktionieren der Produkte […]
Referententwurf auf Seite 1
Verlauf des Verordnungsverfahrens
Der Referentenentwurf wurde den Interessensverbänden und zuständigen Stellen zur Stellungnahme übermittelt. Nach Auswertung der Rückmeldungen wurde im Mai 2024 dann eine neue Fassung der MPBetreibV dem Bundesrat zur Abstimmung vorgelegt, die im Juli – mit einer Anlage zusätzlicher Änderungen – vom Bundesrat beschlossen wurde.
Der Beschluss warf jedoch die vom BMG angestrebte Neuregelung zur Aufbereitung und Weiterverwendung von Einmalprodukten komplett um. Das BMG beabsichtigte, zwei von der EU vorgesehene und auf nationaler Ebene genehmigungspflichtige Aufbereitungsverfahren ausdrücklich in die Verordnung aufzunehmen. Allerdings enthielt der Beschluss des Bundesrates nur noch eine Art des Aufbereitungsverfahrens, was auf erhebliche Kritik stieß.
Angesichts dieser Kontroversen entschied das BMG, die Verordnung vorerst nicht zu verkünden. Stattdessen setzte es sich erneut mit der Änderung der MPBetreibV auseinander und legte einen neuen Referentenentwurf (07.10.2024) vor, der insbesondere eine Überarbeitung von § 9 zur Aufbereitung und Weiterverwendung von Einmalprodukten umfasst. Auch dieser Entwurf wurde erneut den Ländern und Interessensverbänden zur Stellungnahme übermittelt.
Die überarbeitete Verordnung soll im Februar 2025 dem Bundesrat zur Abstimmung vorgelegt werden, mit einem voraussichtlichen Inkrafttreten im März 2025.
Art der Änderungen der Fassung aus Mai 2024 und ihre Auswirkungen auf die Hersteller
Entsprechend dem Anlass der Änderungen betreffen viele der geplanten Änderungen die Software. Damit sind zumindest indirekt auch die Hersteller dieser (Medizinprodukte-)Software betroffen.
| Artikel | Änderung | Auswirkung auf die Hersteller |
| § 4 | Für jede Installation von Software-Aktualisierungen, die die Handhabung der Software nicht nur geringfügig ändert, wird eine Einweisung verlangt. (Geringfügige Änderungen sind z. B. minimale grafisch-gestalterische Anpassungen der Benutzeroberfläche oder für den Benutzer nicht bzw. nur gering wahrnehmbare Fehlerbehebungen oder Sicherheitspatches.) | Die Hersteller müssen Änderungen kommunizieren und die Installation von Updates mit den Betreibern abstimmen. Sie sollten den Betreibern auch Unterlagen für die Einweisung in die „ordnungsgemäße Handhabung“ bereitstellen oder die Einweisungen selbst durchführen. |
| § 7 | Angemessene Maßnahmen zur Instandhaltung müssen auch im Anschluss an sicherheitsrelevante Vorkommnisse (bspw. im Hinblick auf vernetzte Produkte Angriffe auf das Netzwerk), die die ordnungsgemäße Funktionsfähigkeit eines Produktes in Frage stellen können, durchgeführt werden. | Hersteller sollten Hilfestellungen geben, wie die Betreiber das korrekte Funktionieren der Software nach einem Angriff prüfen können. |
| § 7 | Für Software wird klargestellt, dass ihre Instandhaltung auch die Installation verfügbarer sicherheitsrelevanter Software-Updates umfasst. Das kann bspw. dann der Fall sein, wenn das Update Sicherheitspatches beinhaltet. |
Hersteller sollten sicherstellen, dass die Betreiber über verfügbare sicherheitsrelevante Software-Updates informiert werden, sofern diese nicht direkt durch den Hersteller installiert werden. |
| § 17 (neu) | Für bestimmte Software (Software als Medizinprodukt der Klassen IIb und III sowie Software als In-vitro-Diagnostikum der Klassen C und D) muss der Hersteller oder eine vom Hersteller dazu befugte Person die ordnungsgemäße Installation der Software überprüfen sowie eine spezielle Einweisung der Anwender in die sachgerechte Anwendung und den Betrieb der Software-Produkte vornehmen.
Außerdem wird verlangt, dass spätestens alle zwei Jahre angemessene IT-Sicherheitsüberprüfungen der Software-Produkte nach den allgemein anerkannten Regeln der Technik durchgeführt und dokumentiert werden müssen. |
Hersteller müssen die ordnungsgemäße Installation der Software-Produkte sowie die Einweisung der Anwender durchführen.
Hersteller können die IT-Sicherheitsüberprüfungen nur eingeschränkt unterstützen, da diese das Zusammenspiel zwischen der Software und der IT-Infrastruktur der Gesundheitseinrichtung berücksichtigen müssen. |
Änderungshistorie
- 2025-02-13: Artikel weitestgehend überarbeitet und im Hinblick auf neue geplante Änderungen aktualisiert
- 2024-05-25: Boxen mit Warnungen und weiterführenden Informationen in den Kapiteln 1 a) und 2 a) ergänzt
- 2023-11-29: In Kapitel 7 c) die geplanten Änderungen an der MPBetreibV aufgenommen
- 2023-05-04: Artikel aktualisiert, FAQ ergänzt, Verweise auf die alte MPBetreibV korrigiert
- 2021-05-31: Artikel weitgehend überarbeitet, Historie gelöscht, Kapitel 3 eingefügt, Kapitel 6 ergänzt


