Die NMPA Usability Guidance betrifft viele Hersteller von Medizinprodukten und IVD sowie Hersteller von Kombinationsprodukten, welche ihre Produkte in China in den Markt bringen wollen.
Welche Produkte die NMPA-Anforderungen bezüglich Usability erfüllen müssen und worin diese Anforderungen bestehen, klärt dieser Artikel.
1. Wen die NMPA Usability Guidance betrifft
a) Ein- und ausgeschlossene Produktklassen
Das Guidance-Dokument der chinesischen Zulassungsbehörde NMPA ist seit dem 8. Oktober 2024 anwendbar. Es betrifft vor allem die Produkte der Klassen II und III, auch IVD und Kombinationsprodukte („drug-device combination products“). Hingegen fallen IVD-Reagenzien nicht unter den Anwendungsbereich.
b) Bestandsschutz
Bei der Erneuerung von Zulassungen/Registrierungen besteht die NMPA nicht auf zusätzliche Daten aus dem Usability Engineering.
Hingegen entfällt der „Bestandsschutz“ bei substanziellen Änderungen der Nutzer, der Use Scenarios oder des User Interfaces. Dann müssen die Hersteller zumindest die geänderten „Aspekte“ durch das Usability Engineering bewerten.
Der Fachartikel zur NMPA beschreibt Anforderungen der chinesischen Behörde auch jenseits des Usability Engineerings.
2. Entscheidungen zum Bestimmen der NMPA-Anforderungen
Die Usability-Leitlinie der NMPA verpflichtet die Hersteller zu prüfen, welche Anforderungen ihrer Usability Guidance erfüllt werden müssen. Diese Anforderungen hängen von den Antworten auf folgende drei Fragen ab (s. Abbildung 1):
- Ist das Produkt ein „High Use Risk Medical Device“?
- Ist das Produkt neuartig ist (oder es ein Äquivalenzprodukt gibt)?
- Liegen ausreichend Daten von einem Äquivalenzprodukt vor?
Auf diese drei Entscheidungen gehen die folgenden Abschnitte ein.
2.1 Entscheidung, ob das Produkt ein High Use Risk Medical Device ist
Hersteller müssen zuerst bestimmen, ob ihr Produkt ein „High Use Risk Device“ ist. In diesem Kontext definiert die NMPA drei Risikoklassen (s. Tabelle 1).
| Klasse | Schweregrade möglicher Schäden | Recall Level |
| High Use Risk | Incorrect use may lead to serious injury or death | First |
| Moderate Use Risk | Incorrect use may lead to minor harm | Second |
| Low Use Risk | Incorrect use is unlikely to lead to harm | Third or none |
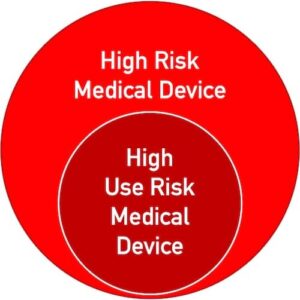
Wichtig ist, dass die Klassifizierung auf den „Use Risks“ basiert, nicht auf den generellen Risken, die das Produkt hervorrufen kann. Allerdings können nur „High Risk Medical Devices“ auch „High Use Risk Medical Devices” sein (s. Abbildung 2).
Für bereits im Markt befindliche Produkte oder den Fall, dass bereits Vergleichsprodukte auf dem Markt sind, kann die Klassifizierung anstatt aufgrund des Schweregrads möglicher Schäden auch auf Basis der „Recall Levels“ durchgeführt werden (s. Tabelle 1).
Insgesamt verlangt die NMPA eine Kategorisierung der mit dem Produkt zu erledigenden Aufgaben („tasks“) gemäß diesen drei Kriterien:
- Risiko („use risk“)
- Dringlichkeit („urgency“)
- Häufigkeit („frequency“)
Nur die erste Klassifizierung (Risiko) entscheidet über die einzuhaltenden Anforderungen. Aber die NMPA erwartet, dass in allen Reports alle Klassifizierungen genannt werden.
Meistens sind High Use Risk Medical Devices kritische IVD bzw. Medizinprodukte, die
- in einer völlig neuen Art genutzt werden,
- eine steile Lernkurve voraussetzen,
- durch nicht-professionelle Anwender genutzt werden oder
- eine hohe Komplexität in der Nutzung haben.
2.2 Entscheidung, ob Produkt neuartig ist
Im zweiten Schritt wird entschieden, ob ein Produkt neuartig ist oder ob es ein vergleichbares Äquivalenzprodukt gibt. Die Äquivalenz bewertet die NMPA anhand mehrerer Charakteristiken (s. Abb. 3).
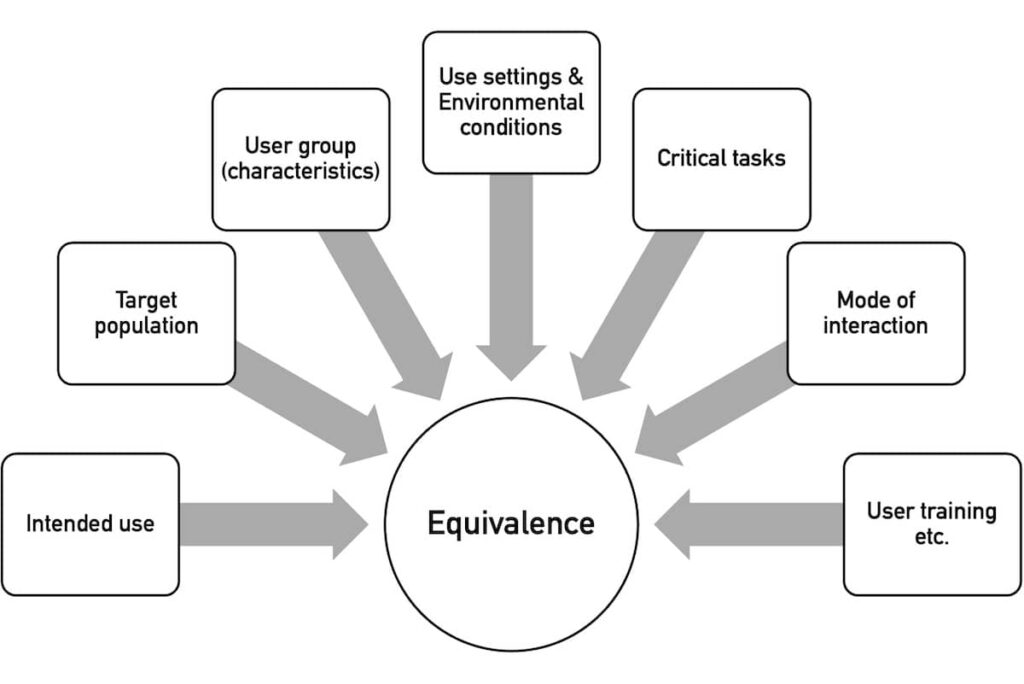
Mit “Use Setting” meint die NMPA die Nutzungsumgebung, z. B. “outpatient clinic, emergency room, operating room, ward, ambulance, home, and public place”.
Unter “Environmental Conditions” versteht sie die physikalische Umgebung, z. B. „space, lighting, temperature, humidity, air pressure, cleanliness, noise, vibration, and radiation”.
Für alle Attribute, die nicht äquivalent sind, müssen die Hersteller entsprechend einen Usability-Test durchführen (siehe Abschnitt c)).
Beim Äquivalenzvergleich dürfen sich die Hersteller nur auf Produkte beziehen, die in China registriert sind und dort vermarktet werden.
2.3 Entscheidung, ob ausreichend Daten vorliegen
Die NMPA besteht darauf, dass der Hersteller die Äquivalenz bezüglich der Usability nachweist, d. h. ausreichend Evidenz dafür liefert. Das sind z. B. folgende Daten:
- Ergebnisse der Usability Validierung des Äquivalenzprodukts
- Falls diese nicht vorliegen, eine Analyse der Post-Market-Usability-Probleme des Äquivalenzprodukts
- Analyse der Post-Market-Usability-Probleme ähnlicher Produkte
Je nach Ergebnis des Vergleichs (siehe Raute „Sufficient Evidence“ in Abb. 2) kann die Durchführung einer Usability Validierung / eines Usability Test in China erforderlich werden. Welche Aktivitäten notwendig sind, beschreibt Tabelle 2.
| Vergleich mit Äquivalenzprodukt | Es gibt keine neuen „Use Risks“ |
Es gibt neue „Use Risks“ |
| Es gibt keine Unterschiede zum Äquivalenzprodukt | Keine Usability-Validierung notwendig | Usability-Validierung für neue Risiken notwendig |
| Es gibt Unterschiede zum Äquivalenzprodukt | Usability-Validierung für die sich unterscheidenden Aspekte notwendig | Usability-Validierung für neue Risiken und für die sich unterscheidenden Aspekte notwendig |
Wenn es Unterschiede zum Äquivalenzprodukt basierend auf den Attributen in Abb. 3 gibt (bspw. eine weitere Nutzergruppe), jedoch keine neuen „Use Risks”, dann muss der Hersteller mit dieser Nutzergruppe einen Usability Test in China durchführen.
In der ersten Zeile von Tabelle 2 wird unterschieden, ob es neue „Use Risks” gibt.
Die Analyse der Post-Market Usability-Probleme ergibt, dass ein Use Scenario, welches bisher nicht mit Risiken verbunden war, aufgrund neuer Erkenntnisse aus dem Markt auf einmal doch risikobezogen ist. Dann muss dieses Szenario einem Usability-Test unterzogen werden.
3. Anforderungen der NMPA Usability Guidance
3.1 Anforderungen ans Usability Testing
Die NMPA spezifiziert in ihrer Usability Guidance viele Anforderungen, die zwar an die der FDA und der IEC 62366-1 erinnern, aber nicht deckungsgleich sind. Beispielsweise fordert sie:
- Durchführung der Tests in China
- Repräsentative Test-Umgebung
- Mindestens 15 repräsentative Vertreter jeder “User Group”
- Testen aller „critical tasks“
- Erfahrene Usability Tester, unter denen keine „Stakeholder“ wie Entwickler sein dürfen
Weder die EU noch die FDA verbieten es explizit, Stakeholder als Tester einzusetzen. Sollte ein Hersteller bei Tests für die NMPA über kein ausreichend großes Team verfügen, kann er auf externe Dienstleister wie das Johner Institut zurückgreifen.
Die NMPA gestattet es generell, auch Daten aus anderen Ländern zu nutzen. Jedoch ist für die Zulassung in China die Zulassung im Heimatland verpflichtend, und die NMPA erwartet
- einen Bericht zu „Post-Market Use Problems“ bei ähnlichen Produkten und
- einen Bericht, der die Unterschiede zwischen den Anforderungen in China und dem anderen Land herausarbeitet sowie spezifiziert, welche Daten noch notwendig sind.
Die NMPA-Leitlinie schreibt dazu explizit:
„detailed data need to be provided to confirm that the differences between China and foreign countries have no significant impact on the user interface validation“ (usability test results from foreign countries).
Da die kulturellen Unterschiede zwischen China und der EU/USA vergleichsweise groß sind, wird es hier besonders schwierig zu argumentieren, warum Nutzer dieselben Fehler machen sollten wie in den westlichen Ländern.
Das erhöht die Wahrscheinlichkeit, dass Usability Tests in China durchgeführt werden müssen.
Die Usability-Tests sind eine der wichtigsten Methoden zur summativen Evaluation, die dem entsprechen, was die NMPA erwartet.
3.2 Anforderungen an die Analysen
Report on Post-Market Use Problems
Der Bericht über bekannte Post-Market-Probleme muss beinhalten,
- nach welchen Informationen gesucht wurde (z. B. zu welchen Produkten),
- wann (Zeitpunkt, Zeitspanne), wie (z. B. Suchbegriffe) und wo (z. B. Datenbanken) gesucht wurde,
- was die Ergebnisse der Suche sind (Quellen, Bewertungen, Schlussfolgerungen).
Report for Comparison
Dieser Bericht ist vergleichbar mit anderen Bewertungen der Äquivalenz, wie man sie von den Predicate Devices der FDA kennt.
Der „Report for Comparison“ sollte auch nennen:
- Das Ziel der Bewertung
- Die verglichenen Produkte
- Die Auswahl des Äquivalenzprodukts
- Daten, welche die Äquivalenz nahelegen
- Die Beschreibung des Evaluationsweges (s. Abb. 1)
- Schlussfolgerungen
- Informationen über die Person, die die Bewertung vorgenommen hat
Usability Test Report
Die Inhalte des Usability Test Reports entsprechen dem, was auch die FDA und die IEC 62366-1 erwarten:
- Test-Objekt (Produkt)
- Probanden
- Testbedingungen, Testumgebung
- Pass-/Fail-Kriterien
- Use-Scenarien und Tasks
- Ergebnisse
- Bewertungen, Schlussfolgerungen
3.3 Anforderungen an die einzureichenden Unterlagen
Abhängig von der Use-Risk-Klassifikation (vgl. Abb. 1) erwartet die NMPA unterschiedliche Unterlagen.
Evaluation Report on Use Errors
Diesen Bericht erwartet die NMPA bei IVD und Medizinprodukten mit niedrigem oder moderaten „Use Risks“. Sie gibt dafür die folgende Struktur vor:
- Basic information
- Level of use risk including justification
- Core elements (e.g. users, use scenarios, and user interfaces)
- Analysis of post-market use problems of similar medical devices
- Use risk management
- Conclusion
Usability Engineering Research Report
Für High-Use-Risk-Produkte müssen Hersteller einen ausführlicheren Bericht erstellen. Die NMPA gibt die folgende Struktur vor:
- Basic information
- Level of use risk including justification
- Core elements (e.g. users, use scenarios, and user interfaces)
- Usability engineering process
- User interface requirements specification
- Use risk management
- Verification and validation of the user interface (include materials described in section 3 b) here, depending on evaluation route)
- Traceability analysis of user interface
- User training protocol
- Conclusion
4. Fazit und Zusammenfassung
Die Anforderungen der NMPA Usability Guidance decken sich in großen Teilen mit den Anforderungen, welche die FDA und die IEC 62366-1 stellen.
Dennoch gibt es einige regulatorische Unterschiede und insbesondere besteht bei Produkten mit High Use Risk die Pflicht, Usability-Tests in China durchzuführen.
Ob die Forderungen der NMPA nur der Patientensicherheit oder auch der Marktabschottung dienen sollen, lässt sich von außen nicht beurteilen.
Erneut stehen die Hersteller vor der Aufgabe, viele marktspezifische Dokumente (insbesondere Berichte) zu erstellen, die in großen Teilen auf denselben Usability-Daten basieren. Hier erweisen sich IT-Systeme als hilfreich, die diese Zulassungsunterlagen auf Knopfdruck generieren.
Das Johner Institut bietet umfangreiche Usability Services an.
- Bereitstellung von Templates für die o. g. Berichte
- Durchführung von Usability Tests in eigenen Usability-Labs in Deutschland und den USA
- Organisation der Durchführung von Usability Tests in Ländern wie China
Das Johner Institut begleitet IVD- und Medizinproduktehersteller auch durch deren digitale Transformation und bietet dazu eine Enterprise-Software für regulatorische Prozesse wie die Zulassung und Marktüberwachung an.
Melden Sie sich (z. B. über die Kontaktseite), um mit einem der Usability Experts des Johner Instituts abzuklären, wie Sie Ihre Produkte schnell und mit minimalem Aufwand in die weltweiten Märkte bringen. Dieses Gespräch ist unverbindlich und kostenfrei.