
Sterben FDA Die Bedeutung der Interoperabilität von medizinischen Geräten, die früh und im Jahr 2017 erkannt haben Anleitung Dokument, Interoperable Medical Devices ‘ veröffentlicht.
Die US -Behörde möchte die Tatsache berücksichtigen, dass die Interoperabilität von medizinischen Geräten für die Gesundheitsversorgung einerseits wichtig ist. Auf der anderen Seite führen Probleme mit mangelnder Interoperabilität zu Risiken.
Dieser Artikel gibt Ihnen einen kurzen Überblick über die Anforderungen der FDA auf “interoperable medizinische Geräte” und gibt Tipps, wie Sie sie treffen können.
1. Über das Leitfadendokument
A. Wenn das FDA -Dokument betrifft
Das Anleitungsdokument “Interoperable Medical Devices” richtet sich an HerstellerSie möchten Medizinprodukte mit Datenschnittstellen in den USA verkaufen. Die Behörde formuliert ihre Erwartungen an die “Zulassungsdokumente”.
Darüber hinaus möchte das FDAMIT gleichzeitig sein eigenes Dokument Inspektoren Vermitteln Sie das Verständnis Ihrer eigenen Autorität zu diesem Thema.
Wie alle “Anleitung Dokumente” ist dies nicht “rechtsverbindlich”. Dennoch erwartet die FDA, dass die Hersteller die in diesem Dokument erforderlichen Informationen für Einreichungen einschließen (z. B. alle Formen von 510 (k), PMA)!
Darüber hinaus wird Herstellern, die in die USA importieren, im Allgemeinen gut empfohlen, allen “Leitlinien” der FDA zu befolgen.
Gleichzeitig bietet dieses Dokument auch Hersteller von Medizinprodukten außerhalb des US -Marktes wertvolle Informationen zur Entwicklung interoperabler medizinischer Geräte.
B. Was die FDA durch Interoperabilität versteht
Die FDA folgt den üblichen Definitionen des Begriffs “Interoperabilität”. Entsprechend Die Fähigkeit von zwei oder mehr Produkten, Technologien oder Systemen, Informationen auszutauschen und ausgetauschte Informationen zu verwenden.
Lesen Sie in diesem Artikel mehr darüber, was Sie unter Interoperabilität meinen und welche Interoperabilitätsniveaus Sie unterscheiden.
2. Anforderungen an “interoperable medizinische Geräte”
Das Dokument, das mit 18 Seiten angenehm kurz ist, führt das Thema in den ersten vier Kapiteln vor und nennt den Bereich der Anwendung und Definitionen.
Die wesentlichen Anforderungen finden Sie in den Kapiteln V und VI:
- Kapitel V gibt Empfehlungen, was bei der Entwicklung interoperabler medizinischer Geräte berücksichtigt werden soll.
- Das Kapitel VI bestimmt, welche Informationen die FDA in Einreichungen erwartet.
Kapitel V: Konstruktionsüberlegungen für interoperable medizinische Geräte ‘
Die FDA fordert, dass die Hersteller beim Entwerfen interoperabler medizinischer Geräte die folgenden Aspekte ansprechen:
A) Zweck der (elektronischen) Schnittstelle
Die Hersteller müssen in einem Zweck bestimmen
- Welche Daten
- In welchem Format,
- Auf welche Weise (Protokolle, Universität/Bidirektional usw.)
- Zwischen welchen Geräten
- Für diesen Zweck (z. B. Getriebealarm, Steuerungsvorrichtung)
- in welchem klinischen Kontext (zB bei Patientenbeatmung)
sollte ersetzt werden.
Die FDA fordert nicht nur das “Zweck“(” Zweck “), aber die Schnittstelle angeben.
B) Vorgeschlagene Benutzer
Die FDA ruft Benutzergruppen wie Kliniker, Servicepersonal, Systemintegratoren, IT und Patienten mit. Diese verfolgen unterschiedliche Ziele bei der Verwendung der Schnittstelle und haben unterschiedliche Kenntnisse.
Hersteller müssen diese Benutzergruppen kennen und im Risikomanagement berücksichtigen.
C) Risikomanagement
Die Hersteller sind verpflichtet, die Risiken aufgrund mangelnder Interoperabilität zu identifizieren und zu beherrschen. Dies beinhaltet Risiken aufgrund falscher Daten oder Befehle, durch Fehlfunktionen der Schnittstellen, durch Probleme mit der IT -Sicherheit oder des Netzwerks (Bandbreite, Latenz usw.).
Hersteller müssen die grundlegenden Sicherheits- und wesentlichen Funktionen sicherstellen (siehe IEC 60601-1).
Darüber hinaus betont die FDA, dass die Hersteller die Risiken über den vollständigen Produktlebenszyklus analysieren und beherrschen müssen (dh nicht nur in der Entwicklung, sondern auch in der Nachmarktphase).
D) Überprüfung und Validierung
Die FDA gibt auch Beispiele für die Berücksichtigung der Hersteller bei „Testen“ (Überprüfung und Validierung):
- Erkennung von korrupten Daten
- Sicherer Betrieb auch für Daten außerhalb von Wertbereichen
- Übergang zum sicheren Zustand
- Konformität mit Interoperabilitätsstandards (falls sie definiert wurden)
- Autorisierung und Authentifizierung
- Benutzerfreundlichkeit
- Auswirkungen auf das Netzwerk und andere Geräte und Systeme
- Die Hersteller sollten diese Tests in simulierten Verwendungsumgebungen durchführen.
E) „Kennzeichnung“
Etiketten, Verwendung und Installationsanweisungen und andere Elemente der “Kennzeichnung” sollten ebenfalls zur Minimierung von Risiken beitragen. Das Leitfadendokument beschreibt genau, was die Kennzeichnung in Kapitel VI Teil D enthalten sollte.
F) „Konsensstandards“
Die FDA -Punkte (empfohlen) auf die “Konsensstandards”, ohne sie direkt im Dokument zu benennen. Sie verknüpft vielmehr die Liste dieser Standards. Beispiele für diese Standards finden Sie unten aufgeführt.
Verwenden Sie dieses Kapitel V, um Ihren Entwicklungsplan zu ergänzen oder nach Vollständigkeit zu überprüfen!
Kapitel VI:-Empfehlungen für Inhalte der Einreichungen vor dem Market ‘
In Kapitel VI beschreibt die FDA, welche Informationen sie erwarten, wenn Sie an “interoperable medizinische Geräte” einreichen. Diese Informationen entsprechen im Wesentlichen der in Kapitel V erwähnten. Daher werden hier nur wenige Besonderheiten dieses Kapitels erwähnt:
- Die Dokumentation sollte: Zweck und Technologien der Schnittstelle, verwendete Standards, Art der Daten ersetzt, funktionale und nicht funktionsfähige Anforderungen, möglicherweise Beschreibung der API
- Die ISO 14971 und die Anforderungen des 21 -CFR -Teils 820.30 (g) müssen beobachtet werden.
- Bei kritischen Anwendungen wie Infusionspumpen müssen alle Testspezifikationen und Ergebnisse eingereicht werden. Bei unkritischen Fall können Zusammenfassungen ausreichen.
- Die Kennzeichnung muss auch den Anforderungen des 21 CFR Teil 801 und 809 erfüllen.
- Die Kennzeichnung (gemeint unter VA ist wahrscheinlich die Anweisungen für die Verwendung) sollte die in Kapitel V in den Unterkapiteln A und B genannten Aspekte beschreiben, die den Benutzern auch die Möglichkeit erhalten, die korrekte Funktionsweise der Schnittstellen zu überprüfen.
Die FDA hat ihr QM -System weitgehend auf ISO 13485 umgestellt. Dies wirkt sich auch auf die Anforderungen des 21 CFR -Teil 820.30 aus. Lesen Sie hier mehr.
3. Erfüllen Sie die Anforderungen
A. Vollständige Dokumente
Das FDA Guidance -Dokument “Interoperable Medical Devices” enthält Anforderungen für die Entwicklung und Dokumentation von medizinischen Geräten. Sie können diese Anforderungen implementieren, indem Sie die folgenden Schritte ausführen:
- Vollständiger Zweck
Stellen Sie sicher, dass der Zweck Ihrer medizinischen Geräte die in diesem Leitfadensdokument genannten Punkte bestimmt, wie z.
– Klinischer Kontext
– Geräte, die verbindlich sind
– Vorgeschlagene Benutzer
– Zweck des Datenaustauschs - Komplement -System-/Softwareanforderungenspezifikation
Vervollständigen Sie die Anforderungen. Geben Sie die Systeme / Software als Black Box an. Verwenden Sie das Blackbox -Modell.
Lesen Sie mehr zu diesem Thema in den Artikeln über das Blackbox -Modell und die Anforderungen
Verwenden Sie nach Möglichkeit Konsensstandards. Weitere Informationen darüber finden Sie weiter unten. - Vollständige Verwendung und Installationsanweisungen
Mit diesen Bestimmungen ergänzen Sie Ihre Anweisungen für Benutzer. Verwenden Sie Kapitel VI D als Checkliste, um die Vollständigkeit zu gewährleisten. - Vollständige Risikomanagementuhren
Ergänzen Sie nun die Risikomanagementuhren. Dies wirkt sich sowohl auf die Risikoetabelle als auch auf den Risikomanagementplan aus, der auch die nachgeschaltete Phase umfassen muss. Passen Sie bei Bedarf den Überwachungsplan nach dem Markt an. Er sollte sich verpflichten, speziell und proaktiv nach Informationen über Risiken durch Interoperabilitätsprobleme zu suchen. - Testprodukt
Führen Sie die Überprüfungs- und Validierungsaktivitäten durch. Die System- oder Softwareanforderungen Spezifikation und Kapitel VI C dienen als Eingabe.
Eine große Herausforderung besteht darin, Systemumgebungen zu schaffen, die für die realen Umgebungen repräsentativ sind.
B. Enthalten “Konsensstandards”
Die FDA hat eine umfangreiche Liste von Konsenusstandards wie folgt veröffentlicht:
- ISO/IEEE 11073–10417 Dritte Ausgabe 2017–04 Gesundheitsinformatik – Personalverwaltung der Personalgesundheit – Teil 10417: Gerätespezialisierung – Glucose -Messgerät.
- ISO/IEEE 11073–10418 Erstausgabe 2014–03–01 Gesundheitsinformatik – Personalverwaltung – Teil 10418: Gerätespezialisierung: International Normalized Ratio (INR) Monitor [including TECHNICAL CORRIGENDUM 1 (2016)].
Wie Sie aus dem Datum sehen können, wurde diese Liste kürzlich aktualisiert.
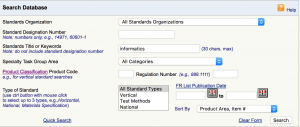
Sie finden diese Konsenus -Standards auf der FDA -Website, auf der Sie das Feld “Informatik” des Schlüsselworts im Feld “Standards -Titel oder Schlüsselwörter” eingeben:
Überlassen Sie die Sicherheit Ihres Patienten nicht dem Zufall
Sicher, es ist sicher mit einem schicksten vom JoHner Institute!
4. Zusammenfassung und Schlussfolgerung
Das FDA -Leitfaden über die “interoperablen medizinischen Geräte” wird seit Jahren fortgesetzt. Trotzdem ist es immer noch nützlich. Auf der FDA -Website “Medical Device Interoperability” finden Sie neuere Gedanken von der FDA zur Interoperabilität.
Es ist irreführend, dass die FDA nur von “legal nicht bindend” und “Empfehlung” spricht. Schließlich erwartet die US -Behörde genau diese in diesem Dokument erwähnten Informationen.
Das Leitfaden dokumentiert nicht “Konsensstandards”, sondern bezieht sich auf die Liste der veröffentlichten. Das macht das Dokument “Warten”. Die Liste der Konsensstandards ist jedoch (auch) auf (physische) medizinische Geräte und die ISO/IEC 11071 -Standards beschränkt. Sie hätten mehr Hilfe für eigenständige Software gewünscht.
Geschichte verändern
- 2025-03-09: Dokument aktualisiert, Kapitelstruktur geändert
- 2017-09-26: Erste Version des veröffentlichten Dokuments


