
Die Koordinierungsgruppe für Medizinprodukte (MDCG) hat ein Leitfaden entworfen, in dem beschrieben wird, wie Hersteller ihre Hersteller haben Medizinprodukte in Klasse 1 MDR-konform.
Das Dokument ist berechtigt Leitnotizen für Hersteller von medizinischen Geräten der Klasse I.. Dieser Artikel fasst dieses Dokument zusammen und gibt den Herstellern dieser Produkte Tipps.
1. Was unterscheidet Medizinprodukte in Klasse 1
a) Klassifizierung von medizinischen Geräten in Klassen
Die medizinische Geräteverordnung MDR unterteilt Medizinprodukte in Klassen 1, 2a, 2b und 3. Gleiches gilt für die Richtlinie für medizinische Geräte MDD. Die Klassen werden oft auch mit römischen Ziffern geschrieben (Klasse I, IIA, IIB und III).
Die Klassifizierungsregeln weisen Produkte mit höheren Risiken für höhere Klassen zu.
Beispiele für medizinische Geräte der Klasse I sind OP -Verachtungen, viele chirurgische Instrumente wie Pinzetten, Latexhandschuhe, Masken, Rollstühle (ohne Motor), Stethoskope, Assoziationen und Brillen.
b) Auswirkung der Klassifizierung
Die Klassen haben keinen (!) Einfluss auf die grundlegenden Sicherheits- und Leistungsanforderungen, deren Hersteller nachweisen müssen. Vielmehr bestimmen die Klassen das Konformitätsbewertungsverfahren, um die Konformität mit diesen Sicherheits- und Leistungsanforderungen zu demonstrieren.
Für medizinische Geräte der Klasse 1 müssen keine benannten Punkte in das Konformitätsbewertungsverfahren einbezogen werden. Darüber hinaus besteht der MDR nicht auf der Zertifizierung des Qualitätsmanagementsystems durch einen benannten Bereich in der Klasse 1 Medizinprodukte. Beide sparen Zeit und Kosten.
Eine Ausnahme sind medizinische Geräte in den Klassen 1s, 1R und 1M:
- 1s: Produkte, die in sterilem Zustand auf dem Markt platziert werden
- 1R: wiederverwendbare chirurgische Instrumente (R steht für “wiederverwendbar”)
- 1M: Produkte mit Messfunktion
Für diese “1*-Produkte” müssen die Hersteller die in der Konformitätsbewertung genannten Positionen integrieren.
2. wie medizinische Geräte der Klasse 1 “genehmigt” werden
Das MDCG beschreibt acht Schritte, die Hersteller auf dem Markt durchlaufen sollten.
Schritt 1: Überprüfen und Bestätigung, dass das Produkt ein medizinisches Gerät ist
Der MDR hat die Definition des Begriffs “medizinisches Gerät” von der MDD fast unverändert übernommen. Es ist daher unwahrscheinlich, dass ein Produkt, das zuvor ein medizinisches Gerät war, nicht mehr unter den Bereich des MDR fällt.
Schritt 2: Überprüfen Sie, ob es sich um ein medizinisches Gerät der Klasse 1 handelt
Dieser Schritt ist unerlässlich, da der MDR die Klassifizierungsregeln geändert hat. Zum Beispiel fällt jetzt fast jede eigenständige Software nicht Mehr in dieser niedrigsten Klasse!
Das MDCG -Leitfadendokument, in dem dieser Artikel erörtert wird, gilt nur für medizinische Geräte der Klasse 1, auch wenn die meisten Anforderungen für alle medizinischen Geräte gelten.
Schritt 3.a: Stellen Sie sicher, dass die grundlegenden Sicherheits- und Leistungsanforderungen erfüllt sind
Es ist offensichtlich, dass die grundlegenden Sicherheits- und Leistungsanforderungen des Anhangs, das ich erfüllt werden muss. Das MDCG weist jedoch darauf hin, dass andere Vorschriften wie die Maschinenrichtlinie möglicherweise ebenfalls beobachtet werden müssen.
Es betont die Bedeutung des Risikomanagements und erinnert daran, dass der Nachweis der Anforderungen durch die Verwendung harmonisierter Normen und gemeinsamen Spezifikationen möglich ist.
Schritt 3.b: Klinische Bewertung erstellen
Das MDCG weist darauf hin, wie wichtig der MDR die klinische Bewertung ist und dass der MDR darauf besteht, dass Hersteller
Schritt 3.C: Erstellen Sie technische Dokumentation
Hersteller von medizinischen Geräten der Klasse 1 müssen die technische Dokumentation gemäß den Anhang II und III erstellen. Dies gilt natürlich für alle medizinischen Geräte.
Das MDCG -Dokument reagiert auf einige Änderungen, die der MDR im Vergleich zum MDD eingeführt hat:
- Grund zur Klassifizierung
- Bissis udi-di
- Verweis auf Vorgängerprodukte und ähnliche Produkte
- Verweise auf angewandte und dann gültige harmonisierte Normen und “gemeinsame Spezifikationen”
- Beschreibung des Nachmarktsurveillanzsystems
Schritt 3.d: Anfrage bei der Namens
Wie bereits erwähnt, müssen Hersteller von Klasse 1* einen benachrichtigten Bereich enthalten, der für die entsprechenden Produktklassen akkreditiert ist:
- Geräte in sterilem Zustand: Code MDS 1005
- Wiederverwendbare chirurgische Instrumente: Code MDS 1006
- Geräte mit einer Messfunktion: Code MDS 1010
Hersteller können in der Nando -Datenbank sehen, die für den jeweiligen Code akkreditiert sind.
Schritt 3.e: Bereiten Sie Anweisungen zur Verwendung und Beschriftung vor
Es ist etwas überraschend, dass nur in Schritt 3 die MDCG die für die Verwendung und die Beschriftung angegebene Anweisungen angibt. Schließlich ermittelt ich den Inhalt in Anhang. Die klinische Bewertung erfordert auch Anweisungen zur Verwendung.
In dem MDCG wird erwähnt, dass für medizinische Geräte in Klasse I keine Anweisungen zur Verwendung vorhanden sind, wenn die sichere Verwendung garantiert ist. Das Dokument geht auch in die Sprachen (ohne auf eine Liste der erforderlichen Sprachen) und die Pflicht der Händler, die dazugehörigen Materialien in diesen Sprachen bereitzustellen.
Symbole werden durch harmonisierte Normen und gemeinsame Spezifikationen angegeben. Das Etikett muss klarstellen, dass das Produkt ein medizinisches Gerät ist.
Schritt 4: Überprüfen Sie die Konformität mit den Anforderungen für die Hersteller
In diesem vierten Schritt geht es weniger um das Produkt als um den Hersteller, VA, weil er verpflichtet ist, ein QM -System festzulegen und eine Versicherung abzuschließen.
Das MDCG reagiert nicht auf die neue Verpflichtung gemäß einer Person, die gemäß Artikel 15 verantwortlich ist, sondern bereits in der Einführung.
Schritt 5: Konformitätserklärung erstellen
Es ist nicht überraschend, dass die Anforderung, dass die Hersteller die Konformität mit dem MDR und gegebenenfalls weitere EU -Vorschriften erklären und dass diese Konformitätserklärung in nationale Sprachen, in denen das Produkt angeboten wird, umsetzen muss.
Schritt 6: Bewerben Sie die CE -Markierung
Dieser Schritt ist auch offensichtlich: Die Hersteller von medizinischen Geräten der Klasse I müssen auch die CE-Markierung anbringen, wobei die Anzahl der genannten Bereiche Teil dieses Labels für Produkte der Klasse-1 sein muss.
Schritt 7: Registrierung des Produkts und des Herstellers im Eudamed
Der siebte Schritt ist in diesem Formular neu: Die Hersteller sind verpflichtet, sich im Eudamed zu registrieren, woraufhin ihnen ein “SRN” zugewiesen wird. Das Gleiche gilt übrigens für EU -Vertreter und Importeure.
Dann ist es die Aufgabe der Hersteller, die medizinischen Geräte zu registrieren. UDI-di und die Grundlage von udi-di werden zugewiesen.
Solange das Eudamed nicht voll funktionsfähig ist, müssen die Hersteller oder EU -Vertreter die verantwortliche Behörde (in Deutschland das Bfarm) informieren oder das Produkt registrieren.
Schritt 8.A: Erfassen und bewerten Sie nach dem Marktdaten
Der MDR verpflichtet die Hersteller, ein Überwachungssystem nach dem Markt für das QM-System einzurichten. Für medizinische Geräte der Klasse 1 müssen die Hersteller nach dem Platzieren auf dem Markt, dem Überwachungsbericht nach dem Markt, einen Bericht über die Überwachung erstellen. Die Anforderungen für diesen Bericht sind geringer als bei den regelmäßigen Sicherheitsaktualisierungsberichten (PSUR).
Schritt 8.b: Vigilanz -System
Der MDCG erinnert daran, dass der MDR auch die Hersteller von Produkten der Klasse 1 verpflichtet. Feldsicherheit Korrekturmaßnahmen (FSCA). Solange der Eudamed nicht in Betrieb ist, gehen diese Berichte an die Behörden (in Deutschland Bfarm, Swiss Swissmedic in der Schweiz). Diese Berichte werden später im Eudamed aufgezeichnet.
Das MDCG -Dokument beschreibt die Aufgaben im Fall von FSCAs relativ umfassend, wie dies derzeit (noch) die Regulierung des Sicherheitsplans für medizinische Geräte ist.
Schritt 8.C: Umgang mit nicht konformen Produkten
Der letzte Schritt betrifft, wie man mit nicht konformen Produkten umgeht. Abgesehen von den bereits erwähnten Berichtsverpflichtungen (Schritt 8.b) müssen die Hersteller entsprechende Korrekturen oder Korrekturmaßnahmen ergreifen.

Der schnellste und billigste Weg, um Ihrem Produkt der Klasse I zuzulassen
Mit unserem Video-Training und Dutzenden von Vorlagen erhalten Sie alles, was Sie für eine schnelle und MDR/IVDR-konforme Genehmigung benötigen. Individuelle Onboarding und ein auf Sie zugeschnittenes Projektplan erleichtern es Ihnen, einen Überblick zu behalten und alle erforderlichen Schritte durchzuführen.
3. Was die Behörden nach Produkten der Klasse I überprüfen
Im Gegensatz zu Produkten mit höherer Klasse liegt die Überwachung der Produkte der Klasse I und deren Hersteller in der Verantwortung der Behörden. Diese Behörden, z. B. Handelsbehörden und Regionalräte, erfüllt sich zunehmend. Sie verlassen sich auf Checklisten wie die folgende vorbildliche Liste. Bitte beachten Sie jedoch, dass nicht alle Checklisten gleich aussehen.
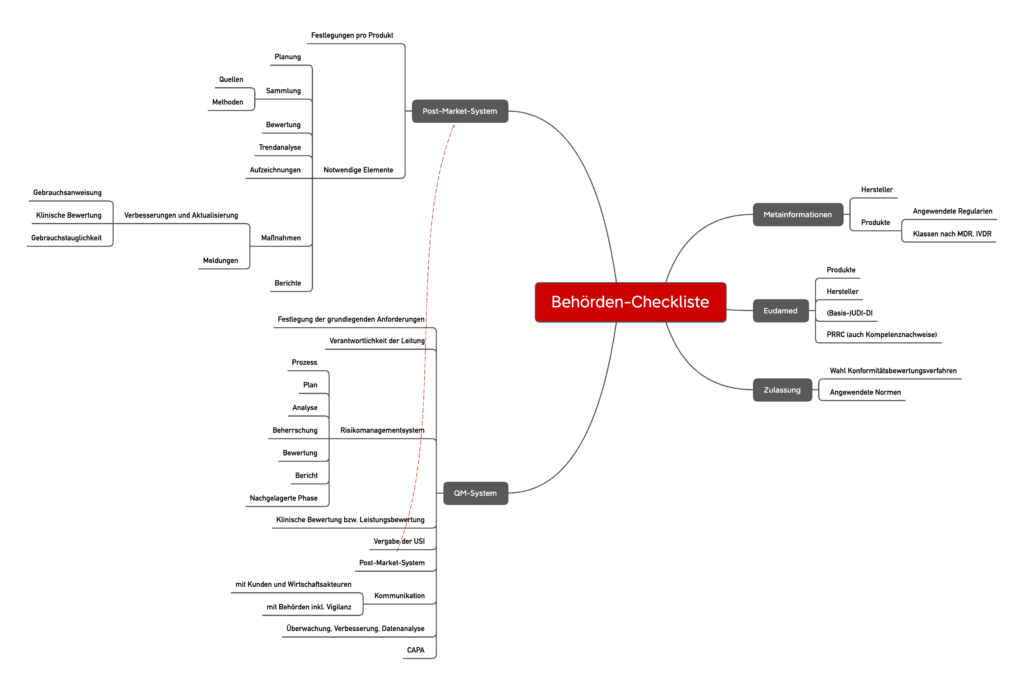
Wie erwartet basieren die Checklisten der Behörden stark auf dem MDR oder dem IVDR. Der Schwerpunkt dieser Checkliste liegt über die Spezifikationen für das Risikomanagementsystem, die Überwachung nach dem Markt und die Wachsamkeit. Hier reagieren die Behörden auf fast jeden Teil der behördlichen Anforderungen.
4. Schlussfolgerung
Das 20-seitige Dokument Leitnotizen für Hersteller von medizinischen Geräten der Klasse I. Sollte – wie der Name schon sagt – als Leitfadenhersteller von medizinischen Geräten der Klasse I dienen.
Das Dokument wird diesem Ziel gerecht. Auf der anderen Seite muss klar sein, dass die darin genannten Anforderungen nicht vollständig und im Wesentlichen nicht spezifisch für die medizinischen Geräte der Klasse 1 sind.
Der Johner -Institut unterstützt Hersteller der Klasse I bei der Erfüllung der Konformität mit den regulatorischen Anforderungen schnell und einfach und somit perfekt für Inspektionen durch Behörden vorbereitet. Machen Sie sich einfach mit uns in Verbindung, z. B. über unser Webformular.
Geschichte verändern
- 2025-03-28: Beispiele in Abschnitt 1. A) eingefügt
- 2023-03-04: Hinweise zu Artikeln zur Software Klasse I ergänzt
- 2021-09-26: Zähler und Kapitel 3 eingefügt.


