
Die Forderung nach „Designverifizierung“ kommt keineswegs nur der FDA zu. In diesem Artikel wird beschrieben, was unter „Designverifizierung“ zu verstehen ist und welche regulatorischen Anforderungen Medizinproduktehersteller erfüllen sollten.
1. Designverifizierung folgt in Kürze
Unter Designverifizierung versteht man die Überprüfung, ob die Entwicklungsergebnisse (Designoutput, z. B. Designunterlagen in Form von Konstruktionszeichnungen) den im Designinput enthaltenen Anforderungen genügen.
Da für diese Prüfungen nicht nur die Dokumente, sondern auch das Produkt benötigt werden, sollte die Konstruktionsprüfung während der Prüfung erfolgen
- konstruktive Phasen (im V-Modell links), beispielsweise in Form von Reviews und
- Analysephasen (direkt im V-Modell) typischerweise in Form von Tests.
Hinweise:
- Die Standards unterscheiden teilweise nicht genau zwischen Stakeholder- und Produktanforderungen und zählen daher gesetzliche Anforderungen (Stakeholder-Anforderungen) als Design-Input, obwohl die Überprüfung der Stakeholder-Anforderungen eine Validierungstätigkeit darstellt.
- Entwicklungsergebnisse in Form von (Architektur-)Dokumenten entstehen häufig im Rahmen mehrerer Entwicklungsaktivitäten und -phasen. Daher kann es zu mehreren Überprüfungen dieser Ergebnisse kommen.
- Die abschließende Prüfung, ob die Designvorgaben in Form von Produktanforderungen erfüllt werden, ist nur auf Produktebene möglich (in Abb. 1 als „Produktsystemtest“ bezeichnet).
Die Herleitung und Diskussion finden Sie im dritten Abschnitt dieses Artikels, häufige Missverständnisse im vierten Abschnitt.
2. Regulatorische Anforderungen
FDA-Anforderungen zur Designverifizierung
Die FDA verlangt in 21 CFR Teil 820.30(f) Folgendes:
Designüberprüfung. Jeder Hersteller muss Verfahren zur Überprüfung des Gerätedesigns einrichten und aufrechterhalten. Die Entwurfsüberprüfung muss bestätigen, dass die Entwurfsausgabe den Anforderungen an die Entwurfseingabe entspricht. Die Ergebnisse der Entwurfsüberprüfung, einschließlich der Identifizierung des Entwurfs, der Methode(n), des Datums und der Person(en), die die Überprüfung durchführt, müssen im DHF dokumentiert werden.
Die „spezifizierten Eigenschaften oder Anforderungen“, die bei einer Verifizierung überprüft werden sollen, beziehen sich daher auf die Designeingaben bei der Designverifizierung. Auch die FDA beschreibt diesen Design-Input:
Design-Input. Jeder Hersteller muss Verfahren einführen und aufrechterhalten, um sicherzustellen, dass die Designanforderungen für ein Produkt angemessen sind und den beabsichtigten Verwendungszweck des Geräts, einschließlich der Bedürfnisse des Benutzers und des Patienten, berücksichtigen. Die Verfahren müssen einen Mechanismus zur Behandlung unvollständiger, mehrdeutiger oder widersprüchlicher Anforderungen umfassen. Die Design-Input-Anforderungen müssen dokumentiert und von einer oder mehreren benannten Personen überprüft und genehmigt werden. Die Genehmigung, einschließlich des Datums und der Unterschrift der Person(en), die die Anforderungen genehmigt, ist zu dokumentieren.
FDA 21 CFR Pat. 820.30 (c)
Wie in einem Artikel über Design-Input geschrieben, entspricht dies in erster Linie der Spezifikation der Produkt-/Systemanforderungen.
ISO 13485-Anforderungen für die Entwicklungsverifizierung
Ganz analog fordert auch die ISO 13485 im Kapitel 7.3.5 („Development Verification“), dass die Entwicklungsergebnisse den Entwicklungsvorgaben entsprechen und dass diese Überprüfung selbstverständlich auch dokumentiert wird. Die ISO 13485 zählt auch „Funktions-, Leistungs- und Sicherheitsanforderungen“ zu den Entwicklungsspezifikationen, die ebenfalls auf dem Niveau der Systemanforderungsspezifikation liegen.
Allerdings listet die ISO 13485 auch rechtliche Anforderungen auf, die Teil der Stakeholder-Anforderungen sind. Eine Überprüfung der Stakeholder-Anforderungen wäre eher eine Validierung.
3. Ableitung
a) Definitionen
Im Gegensatz zur „Designvalidierung“ definiert die FDA den Begriff „Designverifizierung“ nicht explizit. Es definiert jedoch „Verifizierung“.
Bestätigung durch Prüfung und Bereitstellung eines objektiven Nachweises, dass bestimmte Anforderungen erfüllt wurden.
FDA 21 CFR Teil 820.3(aa)
Dies entspricht der Definition der ISO 9000, die auch die Verifizierung beinhaltet Durch Tests mit objektiven Mitteln lässt sich ermitteln, ob bestimmte Eigenschaften oder Anforderungen erfüllt sind.
Gemäß 21 CFR Teil 820.3 und ISO 13485 entspricht die Designverifizierung den (geplanten) Aktivitäten zur Überprüfung (Verifizierung), ob die Designausgabe den Anforderungen der Designeingabe entspricht. Um dies genauer zu verstehen, müssen mehrere Fragen geklärt werden:
- Was ist der Design-Input?
- Was ist das Design und was sind die Design-Outputs?
- Wie sollten Hersteller das Design überprüfen?
Die folgenden Abschnitte geben Antworten.
b) Designeingaben
Die Designeingaben sind in der Regel die Produktanforderungen, insbesondere Anforderungen an die Funktionalität, Leistung, Benutzerfreundlichkeit und Sicherheit des Produkts. Wie oben erläutert, umfasst ISO 13485 auch regulatorische Anforderungen als Design-Inputs.
Regulatorische Anforderungen sind Stakeholder-Anforderungen. Einige regulatorische Anforderungen stellen jedoch spezifische Anforderungen an das Produkt, sodass Hersteller diese Stakeholder-Anforderungen nicht in Produktanforderungen umwandeln müssen.
- Beispiel: IEC 60601-1 legt die maximalen Leckströme eines PEMS fest. Hierbei handelt es sich um eine (überprüfbare) Produktanforderung.
- Gegenbeispiel: Die IEC 62366-1 fordert, dass Hersteller die durch Anwendungsfehler verursachten Risiken minimieren. Hersteller müssen diese Anforderung in konkrete (und überprüfbare) Anforderungen an die Benutzeroberfläche umsetzen.
Der „Practical Guide“ zur ISO 13485 listet weitere Elemente des Design-Inputs auf:
- Erfahrungen mit ähnlichen Produkten
- „Benchmarking-Ergebnisse“
- Ergebnisse von Marktumfragen
- Verpackungsanforderungen
- Standards
c) Design und Designergebnisse
Das Design ist der Entwurf des Produkts. Diese Designaktivitäten führen zu einer Vielzahl von Artefakten:
- Konstruktionszeichnungen
- Stücklisten, Komponentenspezifikationen
- Vorgaben für die Produktion inklusive Produkttests (z. B. Prozesse, Materialien, Werkzeuge)
- Software
- Gebrauchsanweisung, Installations- und Serviceanleitung
Der Praxisleitfaden zählt das Medizinprodukt ausdrücklich zum Designoutput.
d) Designüberprüfung
ISO 13485 verlangt zwei Prüfungen:
- Gemäß Kapitel 7.3.4 sollen die Design-Outputs dahingehend (überprüft und freigegeben) werden, ob sie für die Design-Verifizierung geeignet sind.
- In Kapitel 7.3.6 fordert der Standard „Design and Development Verification“. Ähnlich wie die FDA in 21 CFR Teil 820.30 müssen Hersteller überprüfen, ob der Design-Output den Anforderungen des Design-Inputs entspricht.
Der Zeitpunkt und die Art (Methoden) der zweiten Verifizierung (Design- und Entwicklungsverifizierung) hängen vom zu verifizierenden Gegenstand ab:
- Unterlagen
Einige Entwurfsergebnisse in Form von Dokumenten (z. B. Konstruktionszeichnungen) können unmittelbar nach ihrer Erstellung überprüft werden. Beispielsweise kann eine Systemarchitektur daraufhin überprüft werden, ob sie die in den Produktanforderungen (Design-Input) spezifizierten Schnittstellen bedienen kann. - Produkt, Komponenten
Andere Designergebnisse können nur am Produkt oder seinen Komponenten überprüft werden. Beispielsweise kann die Leistung eines Produkts nur durch das Produkt selbst nachgewiesen werden.
Abhängig von der Art der Prüfung werden unterschiedliche Methoden zur Designüberprüfung eingesetzt:
- Unterlagen: Bewertungen, Checklisten, …
- Produkt, Komponenten: (Bench-)Tests wie Funktionstests, Belastungstests, Simulationen, EMV-Tests, Inspektionen und Methoden der formativen Bewertung wie kognitiver Walkthrough, heuristische Bewertung
4. Praktische Umsetzung
a) Vermeiden Sie Missverständnisse
Hersteller beziehen sich häufig auf ein FDA-Modell (siehe Abb. 2).
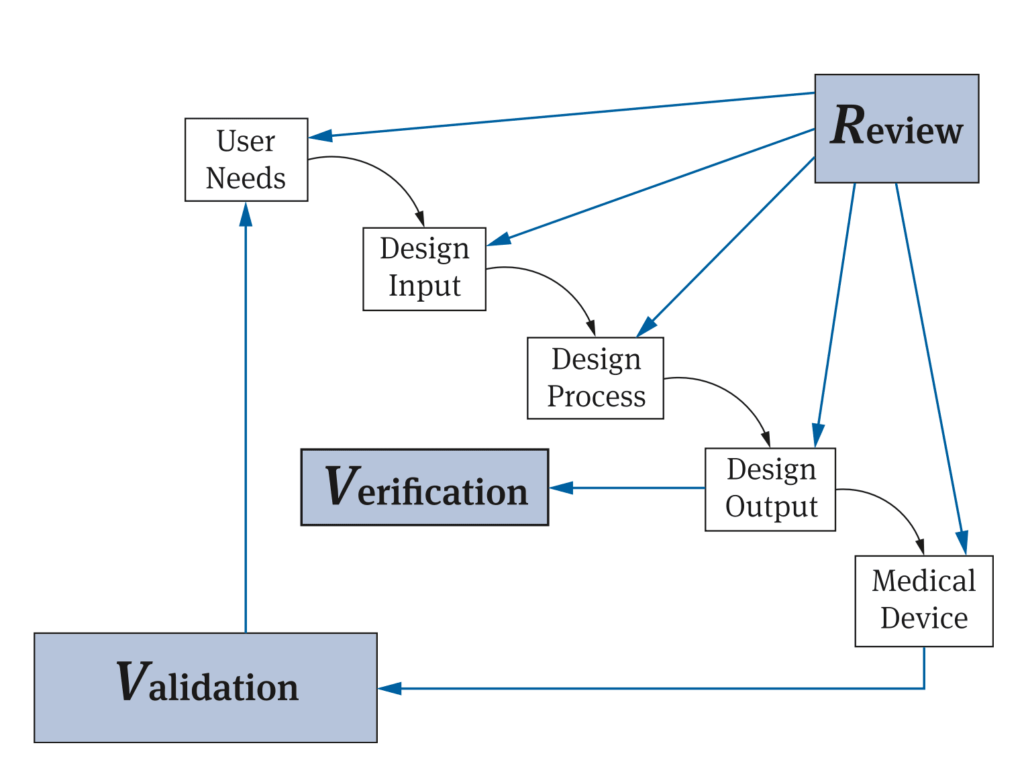
Dieses Diagramm kann jedoch auf verschiedene Weise missverstanden werden:
- Die Designverifizierung erfolgt auf Grundlage des Designoutputs, jedoch nicht unbedingt (erst) am Ende des Entwicklungsprozesses. Dieser Prozess ist normalerweise nicht linear.
- Das Diagramm unterscheidet nicht genau zwischen Aktivitäten und Artefakten. Der „Designprozess“ umfasst beispielsweise Aktivitäten. Der „Design-Output“ sind Artefakte wie Dokumente und Produkte.
- Es scheint, als ob die Überprüfung dazu dient, die Aktivitäten oder Artefakte zu überprüfen. Es muss jedoch zwischen einem „Design Review“ und einer Überprüfung von Zwischenergebnissen unterschieden werden. Beides ist nicht deckungsgleich, wie der Artikel „Design Review ist nicht gleich Design Review“ beschreibt.
- Eine gesetzliche Verpflichtung zur Durchführung einer „Design Review“ für die jeweiligen Tätigkeiten besteht nicht.
- Ein Review ist nur eine von mehreren möglichen Methoden zur Überprüfung von Zwischen- und Endergebnissen.
Nicht jede Überprüfung der Designergebnisse ist eine Designüberprüfung. Eine Designverifizierung muss das Ziel haben, zu überprüfen, ob die „Design-Input-Anforderungen“ erfüllt sind. Eine Überprüfung anhand der Design-Outputs, ob der Design-Prozess eingehalten wurde, käme eher einer Design-Überprüfung im Sinne der ISO 13485 gleich.
b) Beispielsoftware
Wie oben beschrieben, verlangen die Vorschriften, dass die Designverifizierung sicherstellt, dass Entwicklungseingaben wie die Systemanforderungsspezifikation erfüllt werden.
Dies bedeutet, dass die Designverifizierung mindestens die Überprüfung dieser Systemanforderungsspezifikation umfasst. Dies entspricht dem Software-Systemtest.
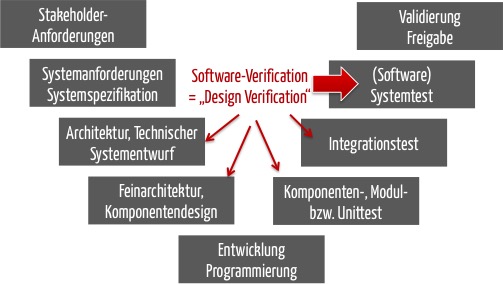
Oft ist es nicht möglich, die korrekte Umsetzung der System-/Softwareanforderungen nur auf Systemebene mit ausreichender „Sicherheit“ zu überprüfen. Genau aus diesem Grund zählen auch die Tests auf den unteren Testebenen zur Designverifizierung.
Streng genommen geht es bei den Software-Architektur-Reviews auch darum, zu prüfen, ob die ausgewählte Planung/Architektur geeignet ist, die „Design-Inputs“ (Software-Anforderungen) zu erfüllen.
Die IEC 62304 spricht explizit von einer Verifizierung der Software-Architektur und nicht von einer Überprüfung der Architektur. Denn eine Überprüfung ist nur eine Möglichkeit für diese Überprüfung. Weitere Optionen sind die Überprüfung von Prototypen, die Bestimmung von Abhängigkeitsmetriken und die Verwendung von Checklisten.
Zusammenfassend lässt sich sagen: Die Designverifizierung für Standalone-Software umfasst
- immer die Softwaresystemtests
- idealerweise auch die Integrations- und Unittests (und statischen Prüfungen des Quellcodes)
- sowie die Verifizierung der Softwarearchitektur.
5. Fazit und Zusammenfassung
Die Designverifizierung wird ebenso missverstanden wie der Begriff Design Review. Dazu tragen auch regulatorische Anforderungen bei, die keinem erkennbaren und weltweit abgestimmten Denkmodell folgen.
In diesem Artikel wurde versucht, dieses Modell bereitzustellen.
Geschichte ändern
- 05.12.2024: Artikel fast komplett neu geschrieben
- 15.10.2015: Artikel zum ersten Mal veröffentlicht


